BCL11 transcription factor A (BCL11A)-related intellectual disability, also known as Dias-Logan syndrome, is a rare autosomal dominant disorder caused by heterozygous pathogenic variants in the BCL11A gene that shows diverse symptoms without developmental regression [1]. The BCL11A gene encodes a zinc finger protein that is predominantly expressed in brain and hematopoietic tissue and works mainly as a transcriptional repressor, which is crucial to the development of the brain and hematopoietic system [2]. Patients with Dias-Logan syndrome present with gross and fine motor delays, growth restriction, intellectual disability, seizures, delayed speech and language development, autism spectrum disorder, and behavioral problems, as well as facial dysmorphism such as hypertelorism, thin upper lip, abnormalities of the external ears, and down slanting palpebral fissures [3-5].
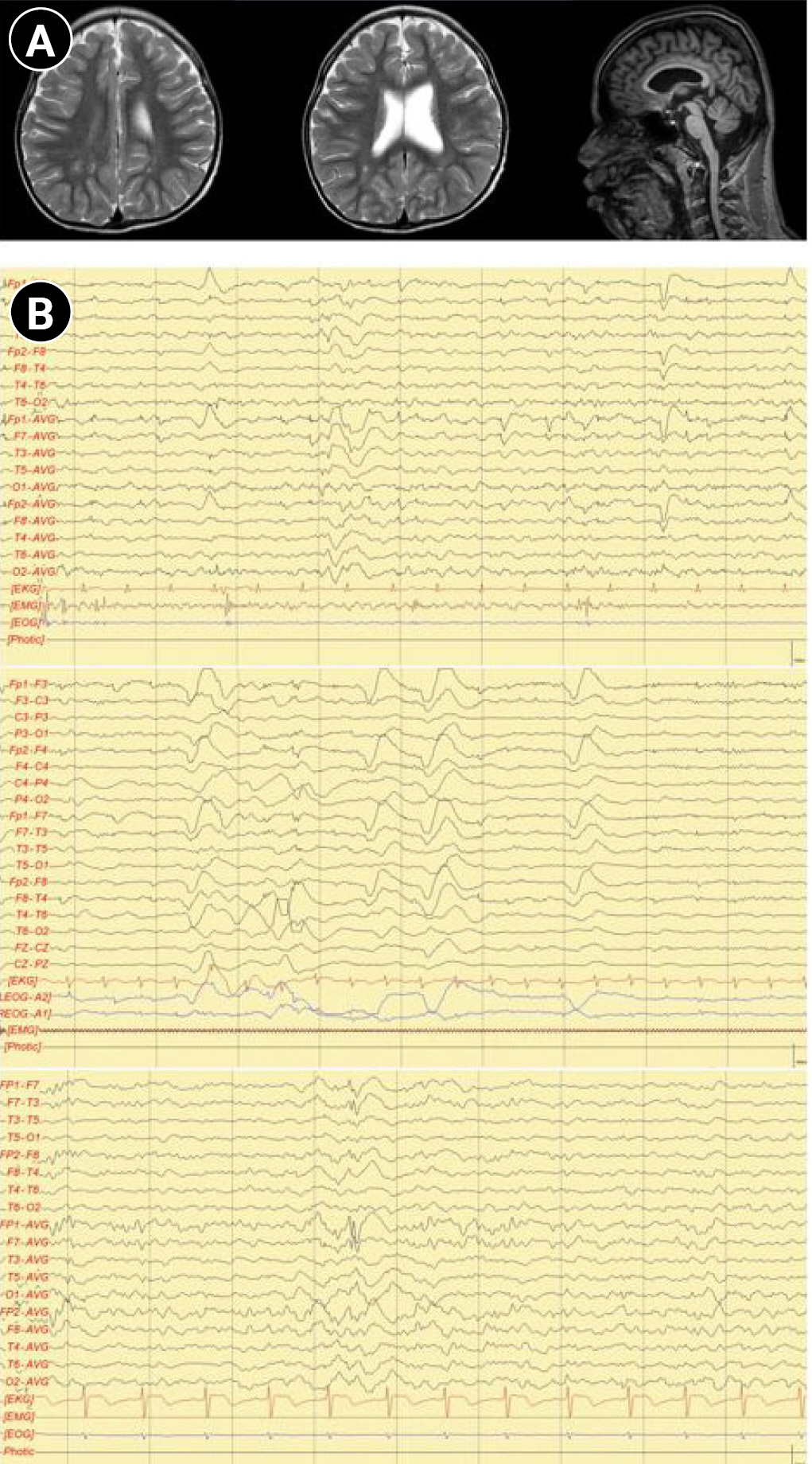
A 17-year-old male patient was brought to the pediatric neurology clinic at 13 months of age with developmental delay. He was able to keep his head steady, starting from 5 to 6 months of age, and could not sit alone or crawl until 13 months. He was born by cesarean section with a birth weight of 2.55 kg and a gestational age of 38 weeks and 4 days. Delays in gross and fine motor skills, as well as language skills, were found. The serum creatine kinase level (149 IU/L; normal range, 26 to 174) and thyroid function test (triiodothyronine, 126.14 ng/dL; free thyroxine, 1.45 ng/dL; and thyroid-stimulating hormone, 3.68 μIU/mL) were normal. A conventional chromosomal study, metabolic screening test using tandem mass screening, and a genetic test for Fragile X syndrome were all normal. A Bayley scale assessment, which was conducted at 14 months of chronological age, showed a developmental age of 6 months in both cognitive and motor skills. He also showed delays in speech and language development. He was not able to speak or pronounce words, even including “mama” and “papa,” at the age of 25 months. A follow-up Bayley test that was performed at 25 months of age still showed severe developmental delays, with a mental developmental age of 7 months and a motor developmental age of 8 months. Brain magnetic resonance imaging performed at 29 months showed mild dilatation of the left lateral ventricle with slightly decreased white matter volume in the peri-trigonal area and thinning of the corpus callosum in the posterior portion (Fig. 1A). He was able to walk independently at 3 years of age. He also showed signs of aggression to strangers, making repetitive low-pitched sounds and inappropriate laughing, and hyperactive motions such as uncontrollable running. He started taking medications for psychiatric symptoms at the department of psychiatry.
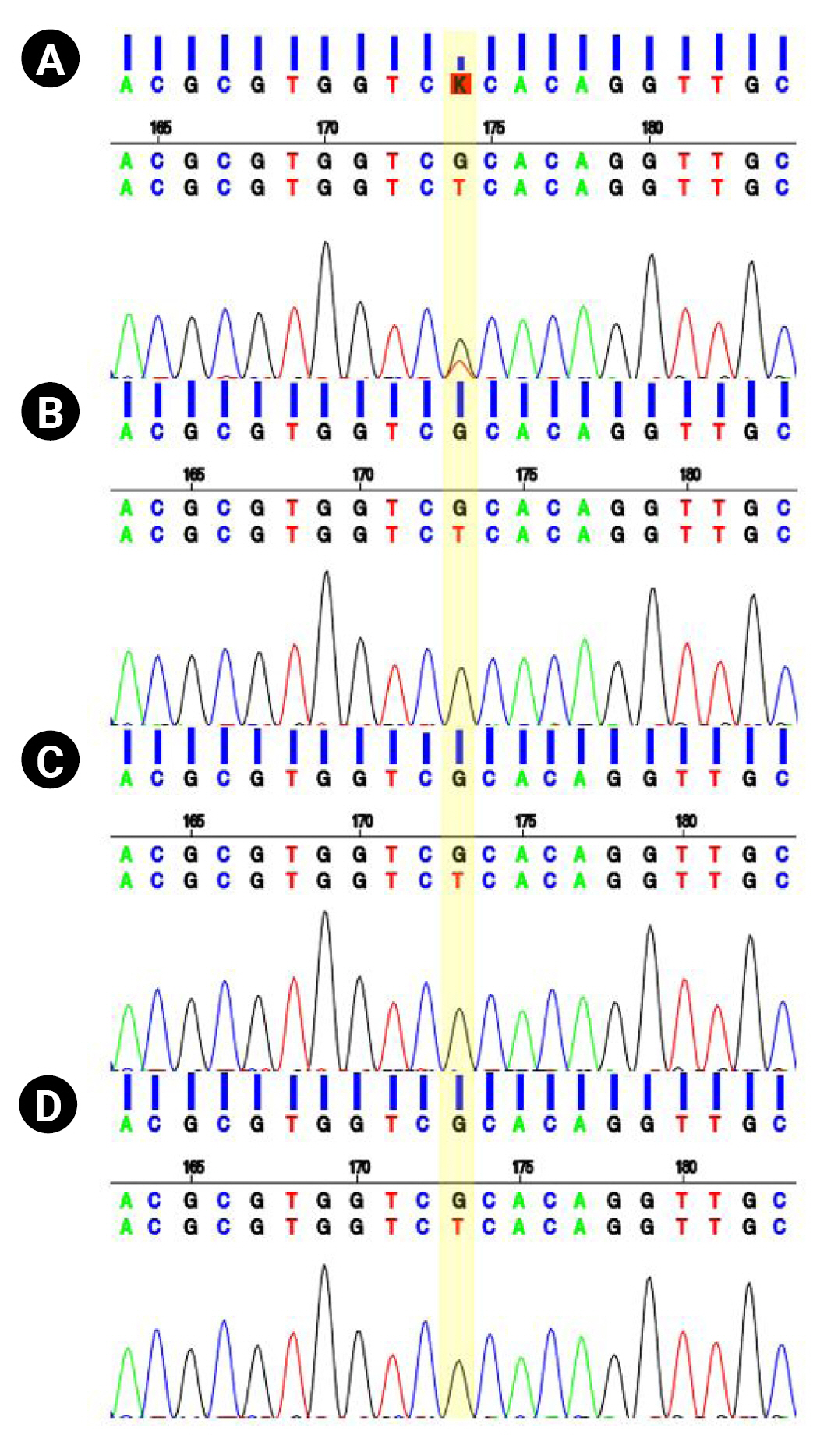
At the age of 13 years, the patient was referred to the pediatric neurology clinic due to recurrent seizures manifested by suddenly falling backward and hyperkinetic movements. On a physical examination, the patient’s facial features showed hypertelorism, downward slanting of both palpebral fissures, arched dark eyebrows, flat midface, thin upper lip and thick everted lower lip, and micrognathia. Electroencephalography showed intermittent bifrontal spike or sharp wave discharges, highly suggestive of focal seizure disorder (Fig. 1B). After adding oxcarbazepine, his seizure episodes declined in number. He has one older brother with intellectual disability. However, his brother did not have a history of seizures. Both parents were healthy, without neurologic problems. Whole-exome sequencing was performed under the impression of epileptic encephalopathy. Genomic DNA was extracted from a buccal swab sample of the patient. All exon regions of all human genes (~22,000) were captured using a Twist Human Core Exome Kit (Twist Bioscience, South San Francisco, CA, USA). The captured regions of the genome were sequenced with an Illumina (San Diego, CA, USA) sequencing machine (Novaseq 6000). The raw genome sequencing data analysis, including alignment to the GRCh37/hg19 human reference genome, variant calling and annotation, was conducted with open-source bioinformatics tools and in-house software (https://pubmed.ncbi.nlm.nih.gov/32901917/). A heterozygous variant was detected in the BCL11A gene (NM_02293.3:c.1230C>A). The substitution generated a nonsense variant at codon 410 in exon 4 (p.Cys410Ter) (Fig. 2), which is expected to cause a loss of normal protein function through nonsense-mediated mRNA decay. It has not been reported in large population cohorts (https://gnomad.broadinstitute.org/). This variant is classified as a likely pathogenic variant according to the recommendation of the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guideline (https://pubmed.ncbi.nlm.nih.gov/25741868/). A segregation study was performed, including the patient’s father, mother, and brother. They did not have the same variant in the BCL11A gene (Fig. 2). He was diagnosed with Dias-Logan syndrome caused by a novel nonsense variant in the BCL11A gene, using whole-exome sequencing. The patient's complete blood count was normal, and the hemoglobin level was 14.6 g/dL. A further assessment of fetal hemoglobin was not performed.
Developmental delay, one of the major chief complaints for pediatric patients, has numerous causes, including birth asphyxia, metabolic diseases, trauma, and specific genetic defects. Seizure is one of the common characteristics that can accompany developmental delay. According to the World Health Organization International Classification of Diseases, 11th revision classification, disorders of intellectual development are a group of etiologically diverse conditions originating during the developmental period characterized by significantly below average intellectual functioning and adaptive behavior that are approximately two or more standard deviations below the mean (approximately less than the 2nd/3rd percentile). The prevalence of intellectual disability was estimated to be about 1% in the United States and other developed countries, with the highest frequency reported in children and adolescents [6].
More than 820 genes are currently known to be associated with diverse childhood neurodevelopmental disorders involving intellectual disability [4]. Remarkable advances in genetics have revealed diverse genes associated with intellectual disability and epilepsy. SCN1A, KCNQ2, ATP1A2, KCNA1, STXBP1, SHANK3, SYNGAP1, CDKL5, SLC2A1, and PCDH19 are some of the genes already known to function at ion channels, synapse, protein kinase, cell metabolism and cell-cell interaction [7]. The transcription factor B-cell lymphoma/leukemia (BCL11A) gene encodes a Krüppel zinc finger protein that regulates transcription by interacting with chicken ovalbumin upstream promoter transcription factor (COUP-TF) proteins and/or sequence-depending DNA binding [1]. Tolve et al. [8] reported that the BCL11A transcription factor is expressed in midbrain dopaminergic neurons in the developing and adult murine brain, and in pluripotent-stem-cell-derived human midbrain dopaminergic neurons. Murine-based studies have shown that the BCL11A gene is a key regulator in neurite arborization, and its proper expression is required for the development of neuronal networks [9]. The BCL11A gene is especially highly expressed in the cortex, caudate, hippocampus, and putamen of the fetal brain [2,8]. BCL11A expression is also high at the embryonic stage, gradually decreasing during development, and inappropriate BCL11A expression may hamper neural network construction, impair cognition, induce intellectual disability, and evoke epilepsy [10]. Dias et al. [4] reported 11 different mutations (three missense and eight nonsense/frameshift mutations) identified in the BCL11A gene. Our patient’s heterozygous variant (c.1230C>A, p.Cys410Ter) in BCL11A was a novel finding. Dias et al. [4] also reported the clinical features of Dias-Logan syndrome, including intellectual disability, language delay, strabismus, flat midface, and thin upper lip among 11 patients. The clinical features of our case were compared with the features previously reported in Dias-Logan syndrome patients (Table 1).
As in our case, previously described patients with Dias-Logan syndrome presented multiple clinical problems such as epilepsy, intellectual disability, and inappropriate behavior. Therefore, a multidisciplinary approach should be implemented, with age-specific supportive care. We also suggest that the BCL11A gene should be considered as a genetic cause of epileptic encephalopathy.
This case was reviewed and approved by the Institutional Review Board of Dankook University Hospital (IRB No. 2022-08-009). The requirement of informed consent for this retrospective study was waived by the board.