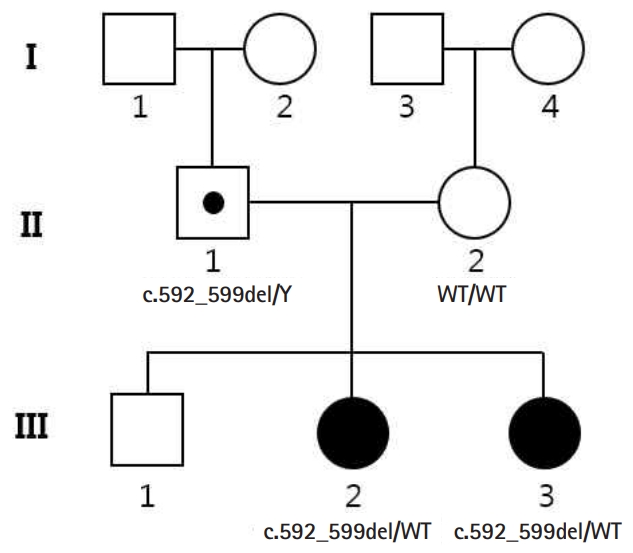
Protocadherin 19 (PCDH19)-related epilepsy is an X-linked epilepsy syndrome characterized by afebrile or febrile seizures, starting in the early years of life, and developmental delay of variable severity [1]. It is caused by mutations in PCDH19 encoding PCDH19, a protein highly expressed during brain development, that is responsible for calcium-dependent cell-to-cell interaction. The disease is of very characteristic heredity; although it is an X chromosome linked disorder, it rarely affects male individuals; this is attributed to “cellular interference,” where the disease could be caused by cell-to-cell signaling between mutated and unmutated cells resulting in hemizygous men being unaffected. Here, we report the case of two Korean female siblings, in which a novel pathogenic variant of PCDH19, inherited from the father, caused different phenotypic expression.
A 23-month-old girl (Sibling 1, III:3) (Fig. 1) was brought to our emergency room after three episodes of generalized tonic clonic seizures with low-grade fever. She had been previously healthy, except for two episodes of simple febrile seizures at 11 and 20 months old, and she had normally reached the developmental milestones without autistic features. She experienced five more seizures on the day of admission, and six times more the next day, even in the absence of fever. The seizures varied in semiology, from generalized tonic clonic seizures lasting 3 to 5 minutes to brief unilateral tonic seizures lasting approximately 1 minute. Unresponsiveness with eye blinking lasting less than 1 minute was also detected. Her brain magnetic resonance imaging was normal, and there was no evidence of central nervous system infection in her cerebrospinal fluid study. She was neurologically intact without deficits and remained alert during the interictal periods, with no more seizures detected after levetiracetam infusion (30 mg/kg). We were informed that she had relevant family history; her 10-year-old sister had epilepsy and her mother had febrile seizures at an early age (Fig. 1). Her sister (Sibling 2, III:2) (Fig. 1) started experiencing febrile seizures at 13 months of age and had clustered brief seizures with or without fever similar to Sibling 1. Administration of multiple antiepileptic drugs was attempted for several years and the frequency of seizures decreased with age. In recent years, she had remained seizure free with levetiracetam monotherapy and her intelligence and social function were within the normal range, but her language skills were relatively delayed. Her mother had simple febrile seizures at an early age, which dissipated at 3 years of age and her intelligence was within the normal range.
The multitude of genes associated with epilepsy and the similarity among clinical phenotypes may complicate diagnosis; hence, we performed whole exome sequencing (WES) with the informed consent of the family members (II:1, II:2, III:2, and III:3). Family-based WES is a very powerful tool for identifying pathogenic variants using expected inheritance patterns. After screening of epilepsy-related genes, we identified a novel deletion variant (NM_001184880.1:c.592_599del) in PCDH19, which was predicted to result in frameshift and premature termination of the PCDH1 protein (p.Arg198Alafs*25) (Fig. 2). The variant was identified as a heterozygote in the two siblings (III:2 and III:3) and as a hemizygote in the father (II:1), but not in the mother (II:2). No other strong candidate genes were located in the WES results. Sibling 1 has remained seizure free for over 6 months with levetiracetam monotherapy (30 mg/kg/day) and has so far normally reached the developmental milestones.
Seizures usually start before 1 year of age, and the presence of fever-sensitive seizures may be clinically similar to the presentation of Dravet syndrome, but seizures in Dravet syndrome usually last longer than 15 minutes (72% of patients) and less than 5 minutes in PCDH19-related epilepsy [1]. Dravet syndrome usually involves clonic or hemiclonic-type seizures, whereas PCDH19-related epilepsy usually involves the focal or hypomotor type. Most importantly, there is a significant difference in prognosis, with remission in adolescence with variable severity of developmental delay being common in PCDH19-related epilepsy, as in our case and severe epileptic encephalopathy often seen in Dravet syndrome [2]. Regarding anticonvulsant treatment, phenytoin is reportedly effective and carbamazepine less so [3]. There have been reports of effective levetiracetam use, including in these siblings, which should be studied further [4]. Fever-sensitive seizures and prominent seizure clustering in varied patterns with short duration (<5 minutes) in girls younger than 1 year, as in our cases, can provide clues for PCDH19-related epilepsy to clinicians [1,4,5].