Mutations in the lysine demethylase 5C (KDM5C) gene are an important cause of the syndromic Claes-Jensen type of X-linked intellectual disability and are associated with mild to severe intellectual disability. Intellectual disability is a clinically variable and genetically heterogeneous disorder characterized by limitations in both intellectual functioning and adaptive behavior, affecting the social and practical skills that are learned before the age of 18 years [1,2]. Intellectual disability affects 1% to 3% of the population worldwide and often coexists with other neurological conditions [3]. Advances in human genetics and clinical research have led to the identification of hundreds of genes responsible for intellectual disability. Numerous studies of large cohorts of patients with intellectual disabilities have shown that the disease has a significantly higher incidence in males, indicating that X-linked gene defects are the main cause of intellectual disability [4]. Here, we report three male siblings with variations in KDM5C, c.1602G>C (p.Trp534Cys), who presented with severe intellectual disability, growth failure, and epilepsy.
The first patient was a 7-year-old boy, who was the first child born to healthy parents with normal intelligence. He was born at 34 weeks of gestation by cesarean section because of congenital intestinal obstruction, and his birth weight was 1,980 g (25th percentile). The patient underwent surgery for duodenal atresia soon after birth. He attained head control at the age of 10 months, creeping at 11 months, rolling at 12 months, crawling at 24 months, sitting alone at 26 months, standing at 36 months, and walking at 46 months. The patient started babbling at the age of 19 months and spoke words after 4 years of age; however, he could not reliably form sentences.
The patient showed global developmental delay on the Korean Bayley Scales of Infant Development-III. At 75 months old, his scores were as follows: cognitive scale, 16 months old; receptive communication scale, 6 months old; expressive communication scale, 10 months old; fine motor scale, 19 months old; and gross motor scale, 18 months old. At the age of 7 years and 5 months, his modified Barthel index score was 26/100. The patient’s full-scale intelligence quotient (Korean-Wechsler Preschool and Primary Scale of Intelligence) was below 40.
The patient also experienced remarkable growth failure. At 6 years and 10 months of age, his height was 95 cm (less than the 3rd percentile), his weight was 12 kg (less than the 3rd percentile), and his head circumference was 46 cm (less than the 3rd percentile). A physical examination revealed marked spasticity in the lower extremities, and ophthalmic screening revealed esotropia and hypermetropia.
The patient also had a history of focal and generalized tonic-clonic seizures since early childhood. Electroencephalography (EEG) revealed a moderately abnormal sleep record due to increased high-amplitude slow-wave activities for his age in the background, which was consistent with non-specific cerebral dysfunction. A variety of anticonvulsants, including phenytoin, levetiracetam, and valproic acid, were used to control seizures, but the epilepsy became intractable to anticonvulsants thereafter.
All biochemical parameters were within their normal range, including lactic acid, pyruvic acid, amino acids, and hormones such as thyroid-stimulating hormone, free thyroxine, and parathyroid hormone. An analysis of urinary organic acids did not reveal any abnormal metabolites. Brain magnetic resonance imaging revealed white matter volume loss with mild ventricular dilatation and diffuse thinning of the corpus callosum (Fig. 1A).
The patient’s second male sibling (6 years old) and third male sibling (4 years old) had a similar clinical course, presenting with severe intellectual disability, global developmental delay, growth failure (height and weight less than the 3rd percentile), and epilepsy. Brain magnetic resonance imaging of the second sibling showed partial agenesis of the corpus callosum (Fig. 1B), and EEG revealed slow and poorly regulated background activity with occasional spike-and-wave discharges from the right centrotemporal area, which was consistent with diffuse cerebral dysfunction and focal seizures.
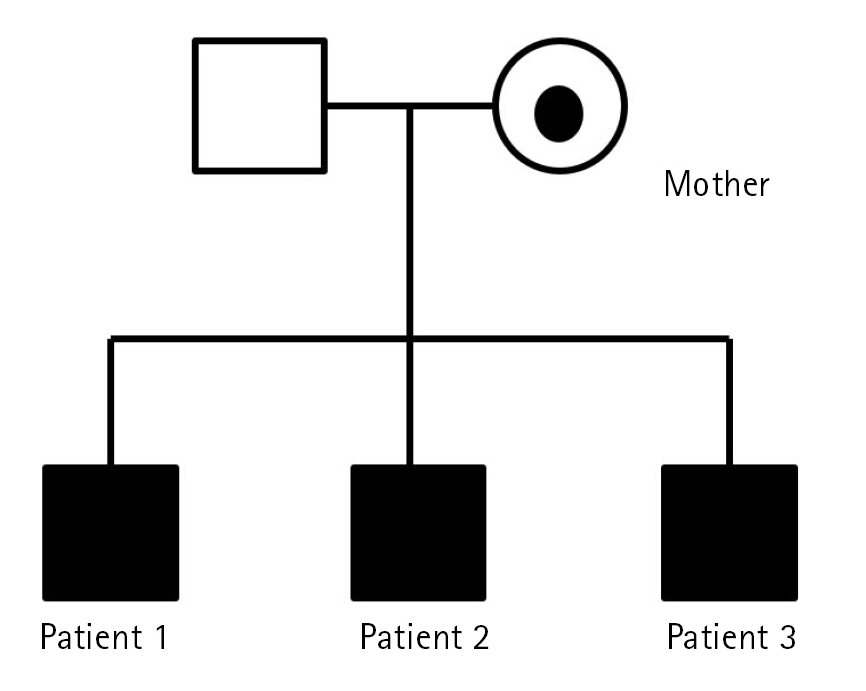
Whole-exome sequencing was performed in the three affected male siblings and the healthy mother (Fig. 2). A hemizygous variant in the KDM5C gene, NM_004187.3:c.1602G>C (p.Trp534Cys), was proposed as the likely pathogenic variant according to the American College of Medical Genetics and Genomics guidelines [5], and their asymptomatic mother was heterozygous for the same mutation. This variant is a missense mutation located in the well-established functional domain of the JmjC domain.
KDM5C variation is inherited as an X-linked recessive trait and is associated with mild to severe intellectual disability. The clinical manifestations in affected males include intellectual disability, developmental delay, delayed speech and language development, autistic behavior, aggressive behavior, seizures, short stature, decreased body weight, microcephaly, progressive spastic paraplegia, lower limb hyperreflexia, and mild dysmorphic features [6]. These clinical features were first described as the Claes-Jensen type of X-linked intellectual development disorder (MIM #300534), and the current understanding of its genetic basis involves a central transcriptional repressive role for KDM5C. The KDM5C gene, located at Xp11.22, encodes a histone demethylase that specifically targets di- and tri-methylated histone H3 lysine ece4 (H3K4me2 and H3K4me3) [7,8] and helps maintain the dynamic balance of the H3K4 methylation state [9]. The H3K4me2/3 substrates are associated with the promoters of transcriptionally active genes and play an essential role in gene transcription [6,7]. In mouse models, loss of KDM5C function caused defective development of the dendrites and dendritic spines, and these defects are often observed in human individuals with intellectual disabilities or autism spectrum disorder [7,10]. In KDM5C-deficient mice, the levels of H3K4me2/3 were increased at the promoters of genes, and KDM5C target genes were aberrantly expressed in these mutant mice [5,7]. KDM5C-deficient mice exhibited fear memory loss, spatial memory defects, increased aggressive behavior, and reduced social preference [7,8,10]. This KDM5C-deficient mice model thereby provided a connection between the dynamic regulation of histone methylation and neurocognitive development. Brooks et al. [1] demonstrated several KDM5C missense mutations with reduced demethylase activity and suggested that the degree of activity reduction correlated with the severity of intellectual disability.
We presented a family of three brothers with X-linked intellectual disability caused by a missense variation (c.1602G>C, p.Trp534Cys) in the KDM5C gene for the first time in Korea. Previously reported patients with missense mutations in the KDM5C gene usually had mild to moderate intellectual impairment, short stature, and microcephaly [6]. However, all three male siblings in this report presented with severe intellectual disability, growth failure, and epilepsy. The oldest brother showed clinical manifestations including global developmental delay, delayed speech and language development, progressive spastic paraplegia, microcephaly, and esotropia, whereas the carrier mother was asymptomatic. The clinical manifestations associated with KDM5C mutations show a degree of variability with regard to neurocognitive disabilities and facial dysmorphism, even in the same family. This variability is probably due to the existence of other unknown disease-causing or compensatory mechanisms related to histone demethylase activity, as well as other epigenetic and environmental factors.
In conclusion, KDM5C mutations are inherited as an X-linked recessive trait and cause a male-specific disorder associated with mild to severe intellectual disability. Therefore, in patients with X-linked intellectual disability, remarkable growth failure, and epilepsy, genetic diseases associated with the KDM5C gene should be taken into consideration. Additional research is needed to identify the genotype-phenotype correlations in the KDM5C gene. Furthermore, the identification of novel disease-causing mechanisms associated with KDM5C could provide new clues to elucidate the genetic causes underlying intellectual disability.
This study was approved by the Institutional Review Board of CHA Bundang Medical Center (IRB No: 2022-07-061). The requirement for written informed consent from the patients was waived by the board owing to the retrospective nature of the study.