Clinical Spectrum and Treatment Outcomes of Patients with Developmental and/or Epileptic Encephalopathy with Spike-and-Wave Activation in Sleep
Article information

Abstract
Purpose
Developmental and/or epileptic encephalopathy with spike-and-wave activation in sleep (D/EE-SWAS) is a spectrum of conditions characterized by various phenotypes of cognitive, linguistic, and behavioral regression associated with spike-and-wave activation in sleep. We aimed to investigate the phenotypic spectrum and treatment outcomes of pediatric patients with D/EE-SWAS.
Methods
We retrospectively analyzed the medical records of pediatric patients diagnosed with D/EE-SWAS and treated at Severance Children’s Hospital from 2006 to 2022. We extracted information from their medical records on electroencephalography before and after treatment, types of treatment, seizure frequency, and developmental profiles. The primary outcome was reduction of the spike-wave index on electroencephalography after treatment.
Results
Twenty-one patients with a median age of 5.3 years (interquartile range, 4.1 to 6.6) at diagnosis were included. Ten patients had delayed development. The patients received various anti-seizure medications. Fourteen received long-term, high-dose steroid therapy, 10 were placed on a ketogenic diet, four received intravenous steroid pulse therapy, and one each was treated with intravenous immunoglobulin and cannabidiol. The most effective treatments were steroid therapy and a ketogenic diet, which were also effective in reducing seizures and improving cognition. Side effects during treatment were transient and treatable.
Conclusion
We described the clinical spectrum of pediatric patients with D/EE-SWAS. Steroid therapy and a ketogenic diet can be considered effective therapeutic options for patients with D/EE SWAS.
Introduction
Developmental and/or epileptic encephalopathy with spike-and-wave activation in sleep (D/EE-SWAS) is a spectrum of conditions characterized by cognitive, linguistic, and behavioral regression associated with marked spike-and-wave activation in sleep, which has been newly defined by the International League Against Epilepsy (ILAE) in 2022. This syndrome includes Landau-Kleffner syndrome (LKS) as a subtype and has been proposed to replace what was formerly named epileptic encephalopathy with continuous spike-and-wave in sleep (CSWS) and atypical benign focal epilepsy of childhood [1].
Electrical status epilepticus in sleep (ESES), an electroencephalography (EEG) pattern of D/EE-SWAS, has no gold-standard treatment and is often resistant to traditional anti-seizure medications [2-4]. ESES is characterized by non-rapid eye movement sleep-induced continuous spike and/or slow waves with a frequency of 1.5 to 3.0 Hz, causing neurocognitive deficits [5]. ESES is originally defined by a spike-wave index (SWI) of at least 85% of epileptiform activity during a particular period [6]. However, some studies have included an SWI in the range of 50% to 85% in the definition of ESES [4]. The clinical presentation of D/EE-SWAS varies, including developmental delay or regression, various cognitive defects such as acquired aphagia, and seizures [6,7]. The age of incidence of D/EE-SWAS ranges from 1 to 14 years, with a peak at 4 to 8 years [1,4]. Severe neurocognitive regression is found at 5 to 6 years [7]. Early detection and intervention are important to address cognitive sequelae that could become permanent [8]. While the pathogenesis of ESES is still unknown, the cortico-thalamic circuitry is strongly suggested to be linked to the epileptiform discharges [7,9]. Moreover, there are some underlying variants in genes such as glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A) that cause ESES [10].
It is difficult to establish a treatment strategy for D/EE-SWAS because there is no accepted first line of treatment, such as anti-seizure medications, immune-modulating therapy, ketogenic diet, or surgery [11,12]. Corticosteroids and adrenocorticotropic hormone are known to be effective in improving cognitive function and reducing the SWI [4,8]. A study on D/EE-SWAS showed that 26 of 37 (70%) children who received steroid therapy had improvement in seizures, and 24 of 36 (67%) patients had cognitive improvement [6]. Another study in 2020 revealed that seven of 10 (70%) CSWS patients had more than 50% seizure reduction with methylprednisolone pulse therapy [13].
This study aimed to investigate the clinical characteristics of pediatric patients diagnosed with D/EE-SWAS. In addition, we evaluated the outcomes of various treatments such as anti-seizure medications, steroid therapy, and a ketogenic diet.
Materials and Methods
1. Study population
This retrospective study was performed at the epilepsy clinic of Severance Children’s Hospital from 2006 to 2022; patients under the age of 18 years diagnosed with D/EE-SWAS were included in the study. The inclusion criteria were as follows: (1) diagnosis of D/EE-SWAS and (2) treatment at Severance Children’s Hospital for at least 3 months. The exclusion criteria were as follows: (1) inability to undergo EEG during sleep and (2) SWI <50% on EEG.
This study was approved by the Institutional Review Board of Severance Hospital (IRB 4-2020-0368). The requirement for informed consent was waived because of the retrospective nature of the study.
2. Data collection
We retrospectively reviewed the patients’ medical records and collected data on the age at D/EE-SWAS diagnosis, age of seizure onset, seizure type, seizure frequency before and after treatment, presence of an underlying disease, findings of brain magnetic resonance imaging (MRI), EEG findings, developmental profiles, treatments (including anti-seizure medications, a ketogenic diet, steroids, and intravenous immunoglobulin [IVIG]), and adverse effects.
We evaluated the EEG findings before and 3 months after each treatment. We performed EEG for 4 hours and ensured that more than 1 hour of sleep state was included in this period. EEGs that did not include over 1 hour of sleep were excluded from the evaluation. In addition, we evaluated the frequency of seizures 1 month before treatment and 3 months after treatment. In order to identify the type and frequency of seizures, guardians were asked to record a seizure diary, and the type and frequency of seizures mentioned in the diary were confirmed at each outpatient clinic visit. Reduction in the frequency of seizures after treatment was evaluated based on the seizure diary. Side effects were also assessed and recorded by guardians while writing the seizure diary. A developmental assessment before the treatment was conducted using questionnaires from an intelligence quotient (IQ) test, and patients were divided into three groups: moderate to severe, mild, and normal. Mild delayed development was determined when the IQ was 70 to 85 or there were two or more domains that were marked “need for follow-up” in the Korean Developmental Screening Test for Infants and Children (K-DST), and moderate to severe delayed development defined as an IQ was less than 70 or two or more domains that were in the “recommendation for further evaluation” category in the K-DST [14]. Two IQ tests were used in this study: the Korean Wechsler Primary and Preschool Scale Intelligence IV (K-WPPSI-IV) for ages between 2.5 and 6 years, and the Korean Wechsler Intelligence Scale for Children (K-WISC-IV) for ages between 6 and 16.9 years.
3. Treatment and outcome assessments
Various traditional anti-seizure medications were administered to patients with D/EE-SWAS. We evaluated the EEG findings and seizure frequency after 3 months of treatment. However, cases in which two or more treatments were administered within 3 months were excluded from the evaluation. All patients were previously on anti-seizure medications, and additional treatment was applied.
Patients who underwent dietary therapy were treated by pediatric epileptologists according to the classic 4:1 or 3:1 ketogenic diet or a modified Atkins diet schedule [15]. Long-term, high-dose steroid therapy was administered according to the schedule presented in our previous paper on Lennox-Gastaut syndrome [16]. During the first 2 weeks of oral prednisolone therapy, 60 mg/day was administered in four divided doses, followed by the same dose of prednisolone on alternate days for 3 months. Intravenous steroid pulse therapy was administered to patients for 3 days at a dose of 30 mg/kg per day (maximum dose, 1 g). IVIG was administered to patients at a dose of 1 g/kg per day for 2 days. Cannabidiol was administered to patients at a dose of 10 mg/kg per day after titration [17].
The primary outcome was the reduction of the SWI on EEG after each treatment. We determined that there was a response to treatment if the SWI value decreased by more than 50% from the pre-treatment value after 3 months of treatment. Additionally, we assessed the reduction in seizure frequency after treatment for patients with seizures. We evaluated treatment as effective when the seizure frequency before treatment was reduced by more than 50%. Changes in development after treatment were evaluated through interviews with guardians in the outpatient clinic. We defined a recurrence as an increase in the SWI by more than 50%.
4. Statistical analysis
Variables with a normal distribution are represented as mean and standard deviation, and variables without normal distribution are represented as medians and interquartile ranges. Data were analyzed using MedCalc version 19.2 (MedCalc Software bvba, Ostend, Belgium).
Results
1. Baseline characteristics
From July 2006 to May 2022, 25 patients were diagnosed with D/EE-SWAS at Severance Children’s Hospital. However, four patients whose EEGs changed to a typical EEG pattern of Lennox-Gastaut syndrome were excluded. Finally, a total of 21 patients were included in this study.
The median age at diagnosis of D/EE-SWAS was 5.3 years (interquartile range, 4.1 to 6.6) and 14 patients (66.7%) were male. In this study, all patients experienced seizures, and the most common type of seizure was focal impaired awareness type (57.1%). Clinical seizure was detected at a median age of 3.7 years (interquartile range, 2.9 to 4.2). One patient showed clinical seizures at the age of 1 month due to a neonatal cerebral hemorrhagic lesion. His EEG findings changed to D/EE-SWAS when he was 3 years old. There were 10 patients with developmental delay, and their developmental delay was recognized by guardians at a median age of 2.8 years (interquartile range, 1.0 to 4.5). Brain MRI results revealed abnormal findings in four patients, including two cases of a mild decrease in hippocampal volume, atrophic changes in the corpus callosum, and chronic hemorrhagic lesions in the thalamus. Diagnostic exome sequencing was performed in 16 of 21 (76.2%) patients, and none of them demonstrated pathogenic variants. The median number of treatments administered to each patient was 4.2 (interquartile range, 3.0 to 5.0), including steroid therapy and a ketogenic diet (Table 1).

Baseline characteristics of patients
2. Effect of each treatment on EEG, seizure, and cognitive improvements
We evaluated the effectiveness of each treatment after 3 months. Each patient had kept the previous anti-seizure medications and received new medication simultaneously. Hence, the efficacy of each treatment was assessed 3 months after each treatment started. Patients were treated with various anti-seizure drugs, long-term, high-dose steroids (66.7%), a ketogenic diet (47.6%), intravenous steroid pulse therapy (19.0%), IVIG (4.8%), and cannabidiol (4.8%). None of the patients underwent epileptic surgery. Most patients (16/21, 76.1%) received valproic acid, and 62.5% demonstrated improvements in EEG and cognitive function. Among patients who were treated with anti-seizure medications, valproic acid showed the most favorable effects (Table 2). However, approximately 70% of patients for whom anti-seizure medications were effective did not maintain the treatment effect for a year, and therefore, other treatments were administered.
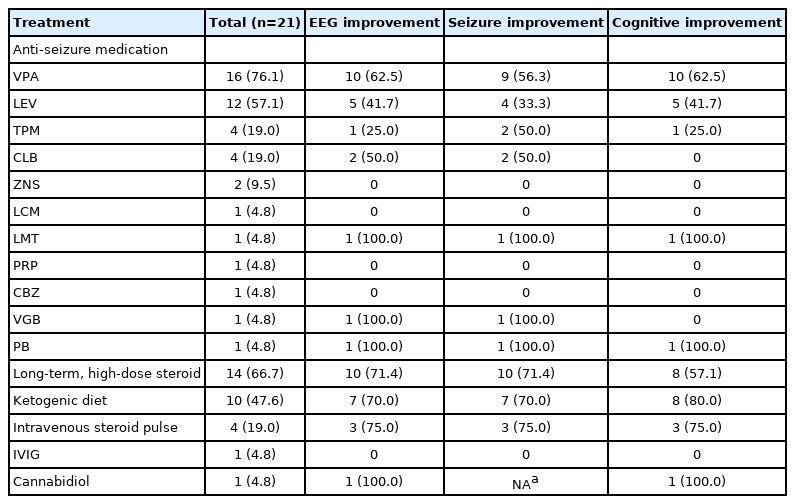
Effect of each treatment on EEG, seizure, and cognitive function
The most effective therapy for EEG improvement was intravenous steroid pulse therapy (3/4, 75.0%). For seizure improvement, intravenous steroid pulse therapy (3/4, 75.0%) yielded the most successful results, followed by long-term, high-dose steroid therapy (10/14, 71.4%). In a previous study, out of 575 cases of ESES patients, high-dose steroid therapy was the second most effective treatment, with 81% (134 of 161) of patients showing improvement, after surgery, which had a 90% improvement rate (56 of 62 patients) [4]. In our study, long-term, high-dose steroid therapy was administered to 14 patients (66.7%); among them, the EEGs of 10 patients (71.4%) showed improvement. Treatment with a 3:1 ketogenic diet was provided to six patients, while two received a 2:1 ketogenic diet and two received a modified Atkins diet. The ketogenic diet had the most favorable effect on cognitive performance (8/10, 80.0%). In addition, the ketogenic diet showed favorable effects on improving EEG and reducing seizure frequency. Approximately 70% of patients who were treated with steroids and a ketogenic diet maintained the therapeutic effect for more than a year. IVIG and cannabidiol were each administered to one patient with intractable epilepsy. Cannabidiol was effective in improving EEG activity. However, this was not observed in the patients treated with IVIG.
Most patients treated with steroid therapy and a ketogenic diet showed more than a 50% reduction in the SWI. The patient presented in Fig. 1 underwent treatment with a long-term, high-dose steroid protocol. This patient not only showed an improvement in the SWI on EEG after 3 months but also demonstrated a dramatic effect on EEG after 6 months of treatment.
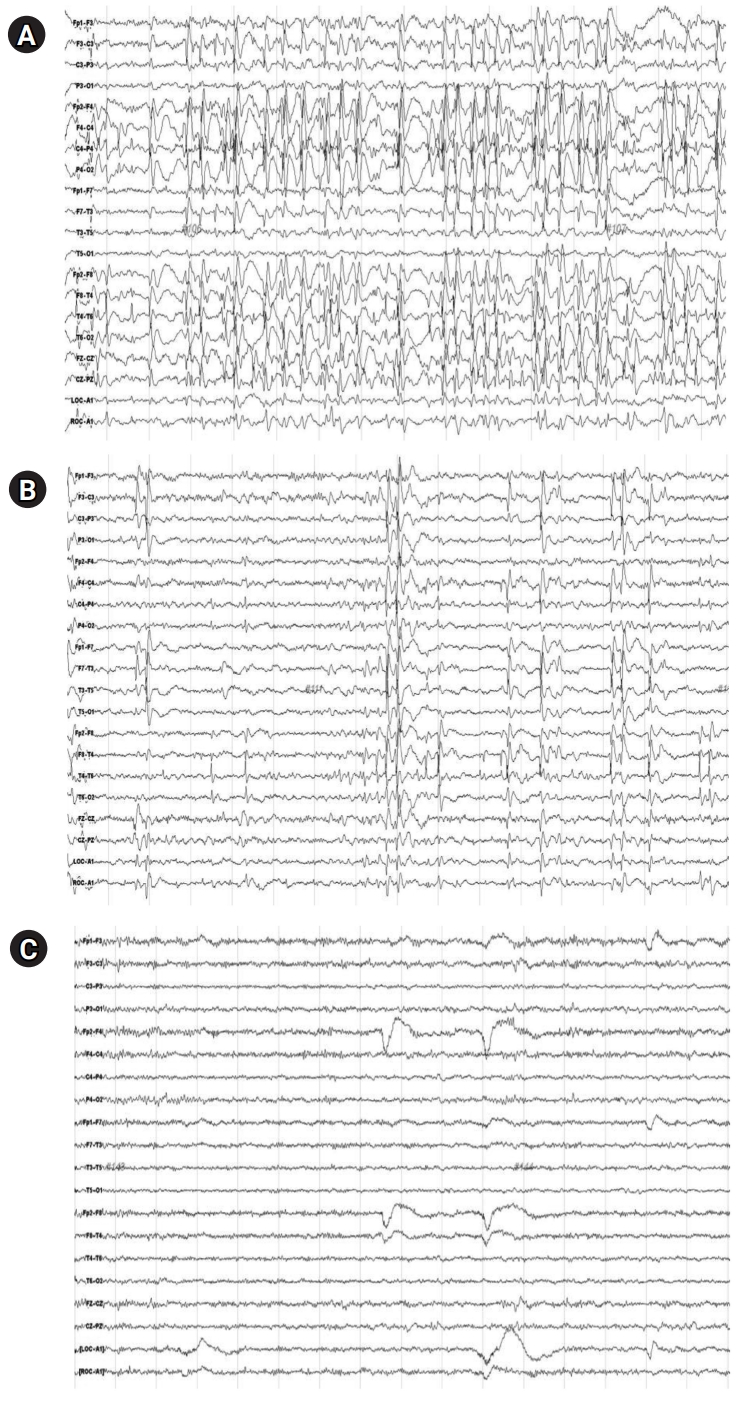
Electroencephalography (EEG) of patient who responded favorably to long-term, high-dose steroid therapy. (A) EEG before steroid therapy. (B) EEG after 3 months of steroid therapy. (C) EEG after 6 months of steroid therapy. A 9-year-old female patient whose disease was intractable with three anti-seizure medications (phenobarbital, valproic acid, and topiramate) received long-term, high-dose steroid therapy for 3 months.
Cognitive improvement was evaluated by a comparison of the findings of the K-WPPSI-IV or K-WISC-IV upon diagnosis with subsequent results from the same tests (13/21, 62.9%). The other patients, who were not tested using the K-WPPSI or K-WISC, were evaluated using the K-DST or based on the guardians’ report. Any improvement was counted, and valproate acid presented the most favorable results, with an improvement rate of 62.5% (10 of 16), except for cannabidiol, which had a 100% improvement rate (one of one).
3. Adverse effects of treatments
Steroid therapy, including long-term, high-dose steroid therapy, and intravenous steroid pulse therapy, showed various side effects in almost all patients. The most common symptoms were weight gain and Cushingoid face. However, most side effects of the steroids were treatable and transient. Among 18 patients, there were no severe side effects such as gross gastrointestinal bleeding, osteoporosis, or cardiovascular effects.
A ketogenic diet was given to 14 patients. However, only 10 patients maintained the ketogenic diet for more than 3 months. The most common side effects of the ketogenic diet were mainly gastrointestinal symptoms, and most of these adverse effects were treatable. Severe metabolic acidosis was treated while controlling the dietary ratio, and there were no life-threatening side effects (Table 3).
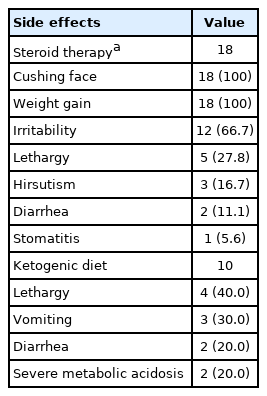
Side effects of patients with steroid therapy and a ketogenic diet
In addition, several patients reported side effects of anti-seizure medications, especially levetiracetam (4/12, 33.3%) and valproic acid (3/16, 18.8%), displaying aggressive behavior or an increase in seizure frequency. Two patients who received clobazam (50%) showed lethargy and general weakness. The single patient who received cannabidiol complained of mild abdominal pain.
Discussion
This study described the clinical characteristics of D/EE-SWAS and evaluated the various treatments employed in its management. The global incidence of D/EE-SWAS is 0.6% to 0.7% of all childhood epilepsies [1]. The exact incidence of D/EE-SWAS in South Korea, is not known. According to the Health Insurance Review & Assessment Service of South Korea, 27 patients with LKS had EEGs showing the ESES pattern and were under medical care in 2019. We can assume that the incidence of D/EE-SWAS is extremely low in South Korea compared to the prevalence of LKS in Japan in 2014, which was 1 in a million in patients aged 5 to 14 years [18].
In this study, the median age of seizure onset in D/EE-SWAS patients was 3.7 years (interquartile range, 2.9 to 4.2), which is similar to that in other studies in which the seizure onset peaked at the age of 4 to 5 years [1]. While the onset of seizures in patients occurred at a median age of 3.7 years, the onset of developmental delay was approximately a year ahead, at a median age of 2.8 years. This suggests that developmental delays could be the early sign of D/EE-SWAS. Other studies have reported that the cause of D/EE-SWAS could be variants in genes such as GRIN2A and structural anomalies of the brain [19,20]. However, our study did not detect any specific pathogenic gene variants or structural anomalies.
While valproate showed a response in nine of 51 (17.6%) CSWS patients in another study in 2021 [6], our study population demonstrated a good response on EEG for nine of 21 (56.3%) patients. However, maintaining the therapeutic effects of anti-seizure medications was a major concern [4]. In Germany, acetazolamide was the most widely used drug, in 96 of 345 patients, resulting in EEG improvements in 22 of 96 (22.9%) patients [6].
In this study, steroid therapy and a ketogenic diet were the key components in lowering the SWI, whereas six recent studies showed variable results of ketogenic diets on ESES [21]. Many other studies have also revealed that steroid therapy yields good outcomes in terms of EEG results and cognitive improvement. There is no standard steroid therapy in terms of steroid type, dose, and treatment duration. In France, among 44 patients with CSWS, including four LKS patients on prednisolone, a favorable response was found in 34 (77.2%) patients; 22 of 44 patients showed improvements on EEG, better development, and shorter SWI duration [22]. A study in China, with 82 ESES patients, including six LKS patients, showed that 68 of 82 (82.9%) ESES patients and six of six (100%) LKS patients had significantly lower SWI after receiving prednisolone (1 to 2 mg/kg/day) for 6 months [23]. However, in another Chinese study, 1-year recurrence was observed in 42 of 82 (51%) ESES patients and three of six (50%) LKS patients. Our study showed a low recurrence rate (approximately 30%) with steroid therapy and a ketogenic diet. Early diagnosis and treatment, with quicker normalization of EEG, could minimize neurocognitive deterioration in patients with CSWS.
The mechanism of corticosteroid therapy in refractory epilepsy remains unclear, although several hypotheses have been proposed. By modulating gamma-aminobutyric acid receptors, steroids reduce the excitability of neurons, thus acting as anti-seizure medications. Other receptors such as glycine, nicotinic acetylcholine, and 5-hydroxytryptamine 3 (5-HT3) receptors, are also known as target-modulating receptors of steroids [24]. A ketogenic diet is known to have numerous mechanisms that together reduce neuronal excitability [25]. For example, medium-chain fatty acids have their own independent anti-epileptic effects, and decanoic acid can act as an anti-seizure medication by directly inhibiting α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors.
This study has several limitations. First, a small number of patients was included because of the low prevalence of D/EE-SWAS. Second, the developmental status was assessed using IQ or the K-DST when the patient was first diagnosed or manifested worsening of the disease. Accordingly, the patient’s cognition after 3 months of each treatment was subjectively evaluated by a caregiver's report, and formal IQ tests were not performed every time. Further studies should use standardized rating scale such as the Clinical Global Impression or a customized questionnaire with a scale given before and after treatment. Third, since cannabidiol and IVIG treatments were received by one patient each, we were not able to properly evaluate their efficacy. Although valproate was used for the first treatment, the order of subsequent treatments was not consistent. Therefore, further research may be needed to determine the effectiveness of the treatment order.
In conclusion, this study demonstrated the characteristics and current treatments of a rare type of epilepsy, D/EE-SWAS, in South Korea. Although the hallmark of D/EE-SWAS is developmental delay, approximately half of the patients had developmental delays and cognitive dysfunction [2]. Steroid therapy and a ketogenic diet could be considered effective therapeutic options for the management of patients with D/EE-SWAS. Further studies should be conducted with larger numbers of patients to compare the efficacy of various treatments.
Notes
Hoon-Chul Kang is an associate editor, Heung Dong Kim, Joon Soo Lee and Se Hee Kim are editorial board members of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: JSL and HCK. Data curation: NT. Formal analysis: DY and HDK. Funding acquisition: HCK. Methodology: HDK, JSL, and HCK. Project administration: HCK. Visualization: DY and SHK. Writing-original draft: NT. Writing-review & editing: DY, SHK, and HCK.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2022R1A2C1012522) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI21C1659) and the Team Science Award of Yonsei University College of Medicine (6-2021-0007).