Clinical and Genetic Spectrum of Tubulinopathy: A Single-Center Study
Article information

Abstract
Purpose
Tubulinopathy represents a group of disorders caused by variants in tubulin genes, which present with a wide spectrum of brain malformations. This study was conducted to provide insight into the phenotypic and genetic spectra of tubulinopathy within the Korean pediatric population.
Methods
Among individuals who underwent genetic testing at a pediatric neurology clinic between June 2011 and December 2021, 15 patients with tubulin gene variants were retrospectively recruited. Clinical features, genetic information, and brain imaging findings were retrospectively reviewed.
Results
The genetic spectra of the patients included TUBA1A (n=5, 33.3%), TUBB4A (n=6, 40.0%), TUBB3 (n=2, 13.3%), TUBB (n=1, 6.7%), and TUBB2A (n=1, 6.7%) variants. Two novel mutations were identified: a c.497A>G; p.(Lys166Arg) variant in TUBA1A and a c.907G>C; p.(Ala303Pro) variant in TUBB. All 15 patients exhibited developmental delays, with a broad spectrum of severity. Other common manifestations included microcephaly (n=10; 66.7%) and seizures (n=9; 60%). A review of the neuroimaging data revealed a range of findings that were both genotype-specific and overlapping across genotypes. In cases of TUBA1A mutation (n=5), four patients (80%) presented with pachygyria and polymicrogyria, while three (60%) displayed cerebellar hypoplasia and dysplasia. All patients with TUBB4A variants (n=6) exhibited hypomyelination, and three (50%) had cerebellar dysplasia.
Conclusion
This study represents the first cohort analysis of tubulin gene mutations associated with tubulinopathy in a Korean pediatric population. It suggests that these mutations can produce a broad spectrum of neurodevelopmental and neuroimaging findings and should be considered within the differential diagnosis in relevant clinical scenarios.
Introduction
Cerebral development requires a highly coordinated sequence of molecular and cellular events to successfully form the brain’s intricate structure, a process that relies heavily on microtubules. These cylindrical, scaffold-like components are crucial for cell division, intracellular transport, and cellular support, making them indispensable for neurogenesis, neuronal migration, and post-migration organization [1]. Microtubules are polymers composed of various tubulin building blocks, including alpha-, beta-, and gamma-tubulin subunits. Tubulin exists in multiple isoforms, each encoded by a different gene. Mutations in these tubulin genes lead to a range of phenotypes, suggesting that each tubulin plays a distinct role in neurons and other tissues. However, the specific molecular mechanisms by which mutations in each tubulin isoform contribute to their associated phenotypes remain poorly understood [2].
Tubulinopathy encompasses a range of disorders resulting from variants in individual tubulin genes, which manifest as a broad array of brain malformations and overlapping phenotypes [2]. Common symptoms include developmental delay, intellectual disability, seizures, microcephaly, and complex cortical development malformations [3]. The discovery of TUBA1A in 2007 as the first known gene associated with malformations of cortical development-type lissencephaly marked the beginning of our understanding in this area. Since then, mutations in other genes that encode alpha-tubulin (TUBA1A), beta-tubulin (TUBB2A, TUBB2B, TUBB3, TUBB4A, and TUBB), and gamma-tubulin (TUBG1) have been identified [4]. While mutations in certain genes, such as TUBA1A, TUBB2B, and TUBB3, are frequently reported in the literature, others (like TUBG1) have been less commonly documented. The advent of next-generation sequencing techniques has led to a rapid expansion in our understanding of the genotype-phenotype spectrum associated with tubulin gene mutations.
Atypical cases are continuously being identified, which is key to achieving phenotypic matching and reaching an accurate genetic diagnosis. Furthermore, this process can yield insights into the mechanisms by which each tubulin isotype modifies the properties of microtubule polymers.
Here, we report the first case series of tubulinopathy in the Korean pediatric population. Our goal was to refine the clinical spectrum of tubulinopathy in relation to genotype. With this study, we aimed to expand the understanding of the genotype-phenotype landscape of tubulinopathy, thereby enhancing molecular testing and diagnosis.
Materials and Methods
Patients who presented with unexplained global developmental delay at the Pediatric Neurology Clinic of Seoul National University Children’s Hospital between June 2011 and December 2021 were considered for this study. Those diagnosed with a tubulin gene mutation via trio whole exome sequencing (WES) were recruited. Trio WES was performed using Illumina technology, with the methods detailed in our previous report [5]. The WES data were aligned with the hg19 reference genome and processed in accordance with the best practices of the Genome Analysis Toolkit. Variant annotation was conducted using the ANNOtate VARiation (ANNOVAR) program, which included the RefSeq gene set and gnomAD, focusing on rare protein-altering variants with a frequency of less than 0.001% in gnomAD. The potential pathogenic impact of the identified variants was assessed using the Combined Annotation Dependent Depletion tool [6].
Patients who lacked parental samples or for whom complete trio samples were not available, precluding the determination of de novo status, were excluded from the study. Variants were evaluated and classified according to the guidelines proposed by the American College of Medical Genetics [7]. The ClinVar and Human Gene Mutation databases were searched for previous reports of variants.
The medical records were retrospectively reviewed, encompassing initial presenting symptoms, age of onset, clinical course, developmental history, current status, physical examination findings, seizure history, and treatment history. All examinations, including electroencephalography (EEG), brain magnetic resonance imaging (MRI), and genetic testing, were thoroughly examined. Patients were categorized based on the tubulin isoforms impacted by mutations and matched with the above information.
The study protocol received approval from the Institutional Review Board of Seoul National University Hospital (IRB No. H-2210-047-1367) and adhered to the applicable guidelines and regulations. To protect confidentiality, any identifiable information of human participants, including patient names, initials, hospital numbers, dates of birth, or other protected healthcare information, remain undisclosed in the publication. Due to the retrospective nature of the study, the requirement for written informed consent was waived.
Results
A total of 15 patients (male:female ratio, 5:10) with tubulin gene variants were identified. The median age at presentation was 12 months (range, 1 to 152). The reasons for presentation included developmental delay (73.3%, n=11), seizures (6.7%, n=1), microcephaly (6.7%, n=1), cyanosis (6.7%, n=1), and feeding difficulty in a neonate (6.7%, n=1).
The median age at genetic diagnosis was 47 months (range, 13 to 163). Fifteen distinct variants were identified across the 15 patients. The genetic profiles of these patients included variants in TUBA1A (n=5, 33.3%), TUBB4A (n=6, 40.0%), TUBB3 (n=2, 13.3%), TUBB (n=1, 6.7%), and TUBB2A (n=1, 6.7%). Two novel mutations were discovered: a c.497A>G; p.Lys166Arg variant in TUBA1A and a c.907G>C; p.Ala303Pro variant in TUBB. These variants were not present in the 1000 Genomes or ClinVar databases (Table 1).
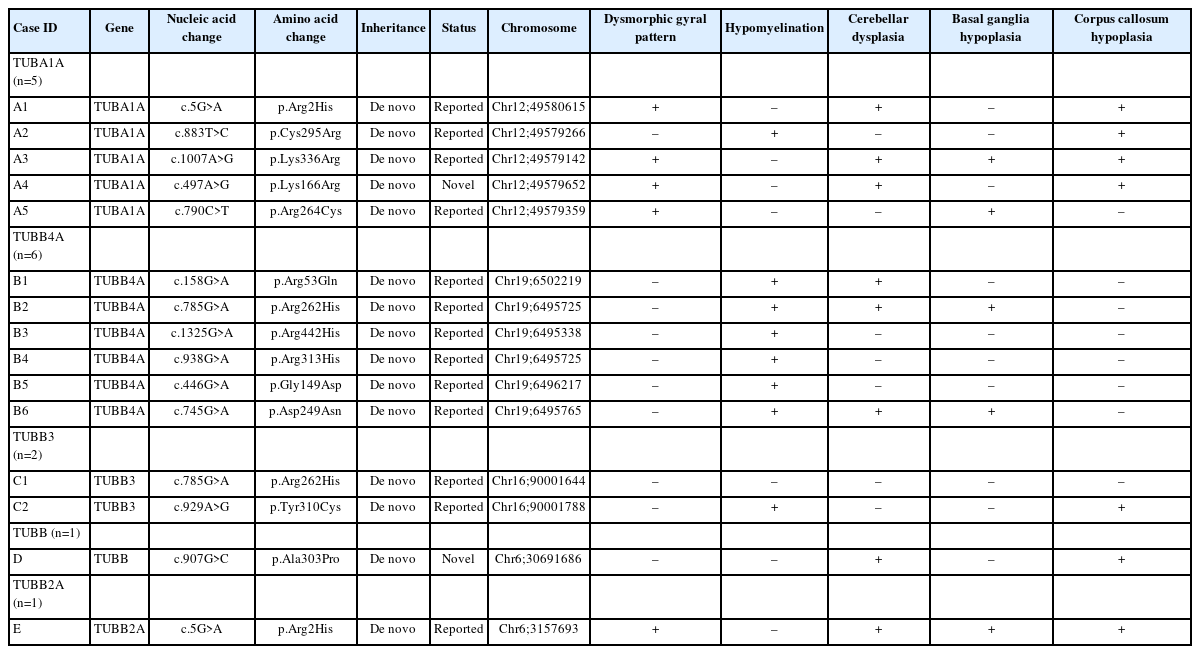
Genotypes and brain imaging findings of individuals with tubulinopathy
The clinical characteristics of the 15 patients with tubulin gene variants are summarized in Table 2. The median age of the patients at the time of review was 5 years (range, 2 to 18). All 15 patients exhibited developmental delays, which varied widely in severity across both motor and language domains. Notably, one patient with the c.1325G>A TUBB4A gene variant (case ID: B3) demonstrated normal intelligence at the age of 6 years. In contrast, a patient with the c.158G>A TUBB4A variant (case ID: B1) lacked expressive language skills at 12 years old and communicated exclusively through nonverbal means. While comprehensive data regarding intelligence quotient were not available, two other patients (case IDs: C1 and D) presented with only mild intellectual disabilities. Developmental regression was uncommon, and significant neurological functional decline was observed in only one patient. That individual, who had the c.745G>A TUBB4A gene variant (case ID: B6), began walking with assistance at approximately 30 months and displayed a wide-based ataxic gait. His walking ability improved gradually until the age of 7 years, when he was able to walk unaided. However, since that time, a gradual increase has been noted in gait disturbance, likely due to increasing spasticity. At present, the patient is unable to stand independently.
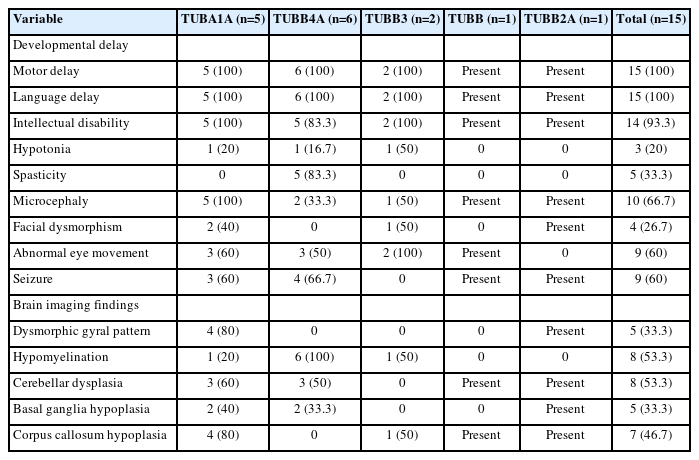
Clinical features
Common clinical presentations associated with mutations in various tubulin isoforms were identified. These include microcephaly (n=10, 66.7%); ocular abnormalities such as strabismus, nystagmus, and optic atrophy (n=9, 60%); and seizures (n=9, 60%), which were observed relatively frequently. Less common features included hypotonia (n=3, 20%) and mild facial dysmorphisms, including short palpebral fissures, upslanting eyes, and low-set ears (n=4, 26.7%). Spasticity was noted in five patients, exclusively among those with a TUBB4A gene variant. The probability of patients with a TUBB4A mutation presenting with spasticity was high, at 83.3% (five of six patients).
A review of patients’ neuroimaging data revealed a broad spectrum of findings, with considerable overlap (Tables 1, 2 and Fig. 1). We identified several structural abnormalities commonly observed across various tubulin isoform mutations. This included dysmorphic gyri such as pachygyria and polymicrogyria (n=5, 33.3%), hypomyelination (n=8, 53.3%), cerebellar dysplasia (n=8, 53.3%), basal ganglia hypoplasia (n=5, 33.3%), and corpus callosum hypoplasia (n=7, 46.7%). Notably, hypomyelination was present in all patients with TUBB4A variants. One patient, who had a TUBB3 variant (case ID: C1), exhibited normal MRI findings. Nine patients who presented with epileptic seizures underwent further analysis in a separate group (Table 3). The median age at seizure onset was 18 months (range, 9 to 52). The types of seizure semiology were diverse, including both focal (n=3, 33.3%) and generalized (n=6, 66.7%) seizures. No semiology was specific to any tubulin isoform, and patients with the same tubulin isoform mutation displayed different types of semiology. For example, each patient with TUBA1A variants (case IDs: A2, A3, and A4) exhibited unique seizure semiologies. Notably, neither of the patients with a TUBB3 variant had a phenotype involving seizures. EEG data from nine patients revealed interictal epileptiform discharges, such as spikes and sharp waves. Of these, six patients also had abnormal background EEG activity.
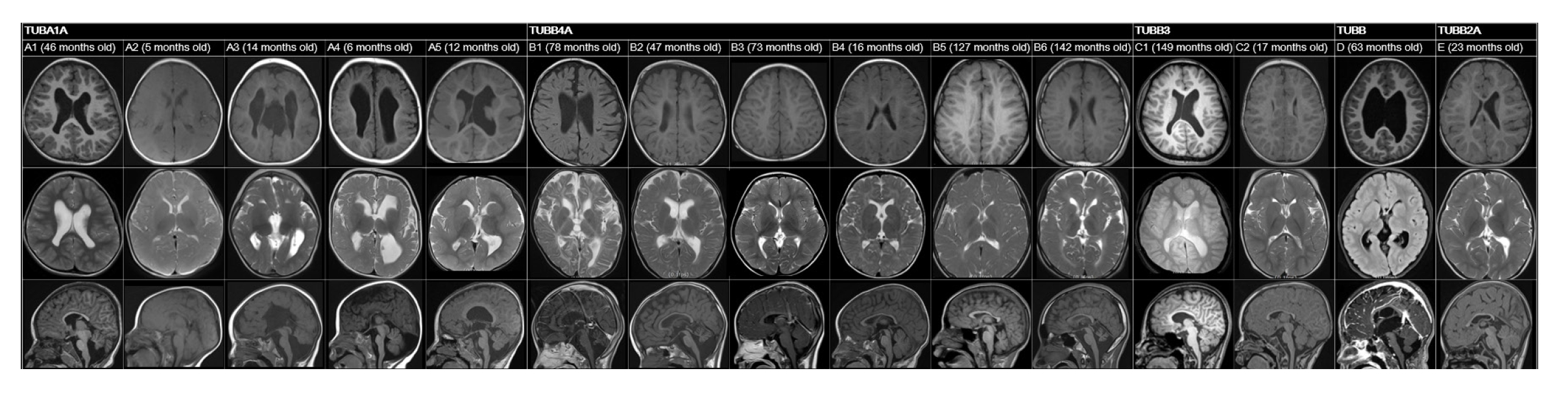
Brain images of individuals with tubulinopathy. Each figure displays T1-axial, T2-axial, and T1-sagittal views arranged horizontally according to case ID.
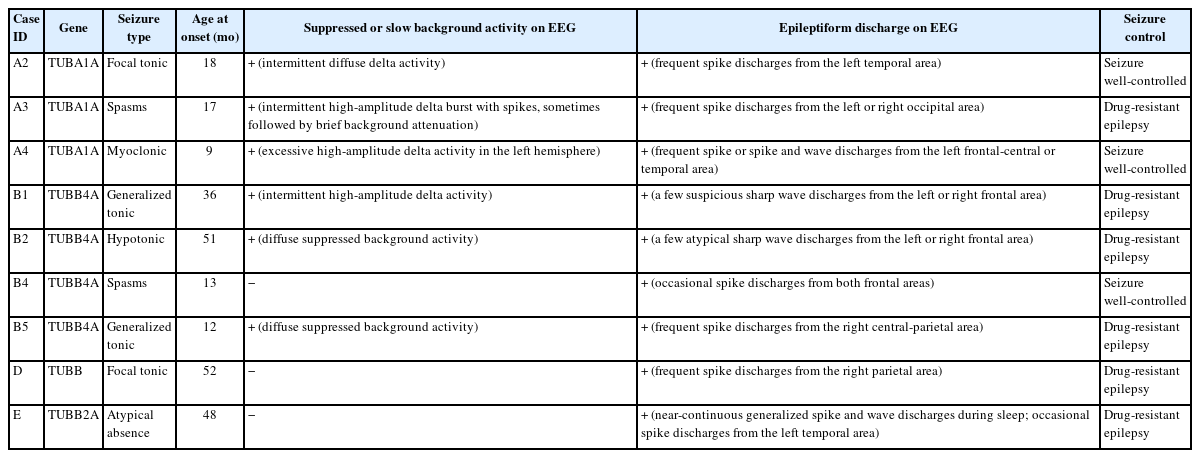
Clinical characteristics of epilepsy in patients with tubulinopathy
Discussion
This study represents the first cohort analysis of Korean pediatric patients diagnosed with tubulinopathies, contributing to the understanding of the genotype-phenotype spectrum. Tubulinopathy was initially characterized by malformations in cortical development. The genotype most often associated with marked structural anomalies is TUBA1A. TUBA1A-related tubulinopathies exhibit prominent features of various cortical malformations, including anomalies in cortical gyration, the basal ganglia, and the corpus callosum [4]. In the present study, among the five patients with TUBA1A variants, dysmorphic gyral pattern (n=4) and corpus callosum hypoplasia (n=4) were the most common findings. Notably, grossly dysmorphic gyral patterns, such as polymicrogyria or pachygyria, were relatively specific to variants of TUBA1A.
Cortical malformations have also been observed in cases involving other genes. Reports on TUBB variants are limited, and the specific phenotype of brain malformation associated with TUBB mutations remains unclear [8]. In this case series, one patient with a novel TUBB variant (c.907G>C; p.Ala303Pro) exhibited a range of anomalies, including asymmetric basal ganglia with a globular appearance, protrusion of the caudate head into the lateral ventricle, thinning of the corpus callosum, and hypoplasia of the cerebellar vermis. Mutations in TUBB3 can result in a spectrum of MRI findings, from normal results to dysgenesis of the corpus callosum, anterior commissure, and internal capsule, as well as a generalized loss of white matter and basal ganglia dysmorphism [9]. Correspondingly, one patient with a TUBB3 mutation in the present report had completely normal MRI findings. Patterns of cortical malformation—particularly abnormal gyral patterns—are often linked to TUBA1A mutations, while hypoplasia or dysplasia of the cerebellum, basal ganglia, or corpus callosum are less specific indicators.
Tubulinopathy may also manifest as hypomyelination, which should be considered in the differential diagnosis of hypomyelination. Mutation of the TUBB4A gene is linked to hypomyelinating leukoencephalopathy and the condition known as hypomyelination with atrophy of the basal ganglia and cerebellum [10]. Consistent with prior research, all patients in our study with TUBB4A mutations (n=6) exhibited hypomyelination, and half of these individuals also had cerebellar atrophy. While TUBB4A mutations are closely associated with hypomyelination, they are not the sole cause; other genotypes, including TUBA1A and TUBB3, were also found to be associated with hypomyelination in our cohort. The presence of hypomyelination should prompt consideration of tubulinopathy as a diagnosis, particularly when accompanied by other structural brain abnormalities.
Tubulinopathy is characterized by developmental delay, which was evident in all patients in this study. However, the severity of the delay varies widely, even among individuals with the same gene mutation. In our study, a patient with a TUBB4A mutation featuring the c.1325G>A variant (case ID: B3) demonstrated near-normal intelligence, despite the presence of hypomyelination observed in brain imaging. While developmental delays are a universal observation in these cases, the degree to which they are experienced can vary considerably.
Patients with tubulinopathy may exhibit additional neurological signs that are key to diagnosis. In a previous study, 23 out of 27 patients with TUBA1A mutations (85.2%) displayed spastic diplegia or quadriplegia, a finding also observed in 30% (three out of 10) of patients with TUBB3 mutations [8]. A 4-year-old boy with the TUBB2A p.Ala248Val variant presented with developmental delay, spastic diplegia, and an exaggerated startle response, but no epilepsy [11]. Notably, in our current cohort, spasticity was observed only in patients with the TUBB4A gene variant, affecting 83.3% (five out of six) of these cases. Although previous reports have suggested that alpha-tubulinopathy is more likely to cause spasticity than beta-tubulinopathy, the variability in our findings—potentially due to the small sample size or ethnic specificity—indicates that nearly any genotype may present with these features. In our cohort, TUBB4A mutation was also the only variant associated with substantial neurological functional regression due to spasticity. Since worsening spasticity can hinder walking, as seen in one patient (case ID: B6), spasticity must be managed proactively through rehabilitation and medication whenever possible.
In contrast, TUBB3-related tubulinopathies cause congenital fibrosis of the extraocular muscles type 3 and other forms of paralytic strabismus [9,12]. In the present study, both patients with TUBB3 variants exhibited abnormal eye movements. However, this feature is not specific to TUBB3, as it was also observed in patients with TUBA1A, TUBB4A, and TUBB mutations. Therefore, while abnormal eye movements are commonly seen in patients with TUBB3 mutations, they can also be present in patients with other genotypes.
In this study, nine patients (60%) presented with epileptic seizures; however, no individual with the TUBB3 gene variant exhibited such symptoms. This observation aligns with prior research, which indicated that among 62 patients with the TUBB3 variant, only three experienced febrile seizures.
In conclusion, we present the first case series of tubulinopathy in Korean patients. Considering the variety observed in brain imaging and clinical phenotypes, tubulinopathy should be consistently included in the differential diagnosis of global developmental delay. Accompanying characteristics, such as abnormalities in brain imaging (including cortical malformation and hypomyelination) and neurological symptoms (such as abnormal eye movements, spasticity, and seizures), may offer clues for the diagnosis of tubulinopathy. While some features were more frequently associated with mutations in specific tubulin isoforms, a notable overlap was observed in most features.
One limitation of the present study was the relatively small number of cases examined. To reinforce the findings, a larger collection of case reports is necessary.
Notes
Ki Joong Kim and Jong-Hee Chae are editorial board members of the journal, but they were not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: HJS, JHC, and WJK. Data curation: HJS, MK, HJK, JSC, SYK, BCL, KJK, JHC, and WJK. Formal analysis: HJS and WJK. Methodology: HJS, JHC, and WJK. Project administration: HJS, MK, HJK, JSC, SYK, BCL, KJK, JHC, and WJK. Visualization: HJS. Writing-original draft: HJS. Writing-review & editing: WJK.
Acknowledgements
The research reported in this publication was supported by the Department of Pediatrics and Rare Disease Center at Seoul National University.