Findings
Triple A syndrome, also known as Allgrove syndrome, was first described in 1978 within a North African family [1]. This condition is extremely rare, with fewer than 200 cases reported to date [2,3]. Allgrove syndrome is an autosomal recessive disorder resulting from a mutation in the AAAS gene, which is located on the long arm of chromosome 12 (12q13). This gene encodes a protein known as ALADIN, an acronym for alacrimia-achalasia-adrenal insufficiency-neurologic disorder [4,5]. The classical clinical spectrum of this syndrome includes alacrimia, achalasia, and adrenal insufficiency (AI). The complete triad is present in approximately 70% of cases [2]. Over time, most patients develop neurological symptoms.
Herein, we report three cases of patients with Allgrove syndrome, with the aim of highlighting the neurological dysfunction observed in this condition. These manifestations have often been overlooked and inadequately described in the existing literature. We conducted a cross-sectional study at the Endocrinology Department of Hedi Chaker University Hospital in Sfax, the economic capital of Tunisia. The study data were retrieved from the medical charts of patients diagnosed with Allgrove syndrome who also exhibited neurological abnormalities. This research received approval from the Medical Ethics Committee of Sfax (approval number 0522/2023). Written informed consent was obtained from the participants in this research.
A 22-year-old man, born to non-consanguineous parents, presented with no notable family medical history. At 3 years old, he had developed hypoglycemia, metabolic convulsions, diffuse hyperpigmentation, and hyponatremia, indicative of Addison disease. This diagnosis was confirmed by observations of low plasma cortisol concentration and elevated adrenocorticotrophic hormone (ACTH) levels. At a follow-up visit, the patient’s mother mentioned that he had never produced tears at any age. Consequently, an ophthalmologic examination was conducted, leading to the diagnosis of alacrima.
The patient also reported severe difficulties with deglutition, dysphagia, and regurgitations consisting of saliva. A radiological study of esophageal transit confirmed achalasia, which was resolved with two balloon dilatations of the esophagus at the ages of 7 and 14 years. Considering these findings, a diagnosis of Allgrove syndrome was suspected and subsequently confirmed when genetic analysis revealed a splice donor mutation in the AAAS gene, specifically IVS14+1G→A.
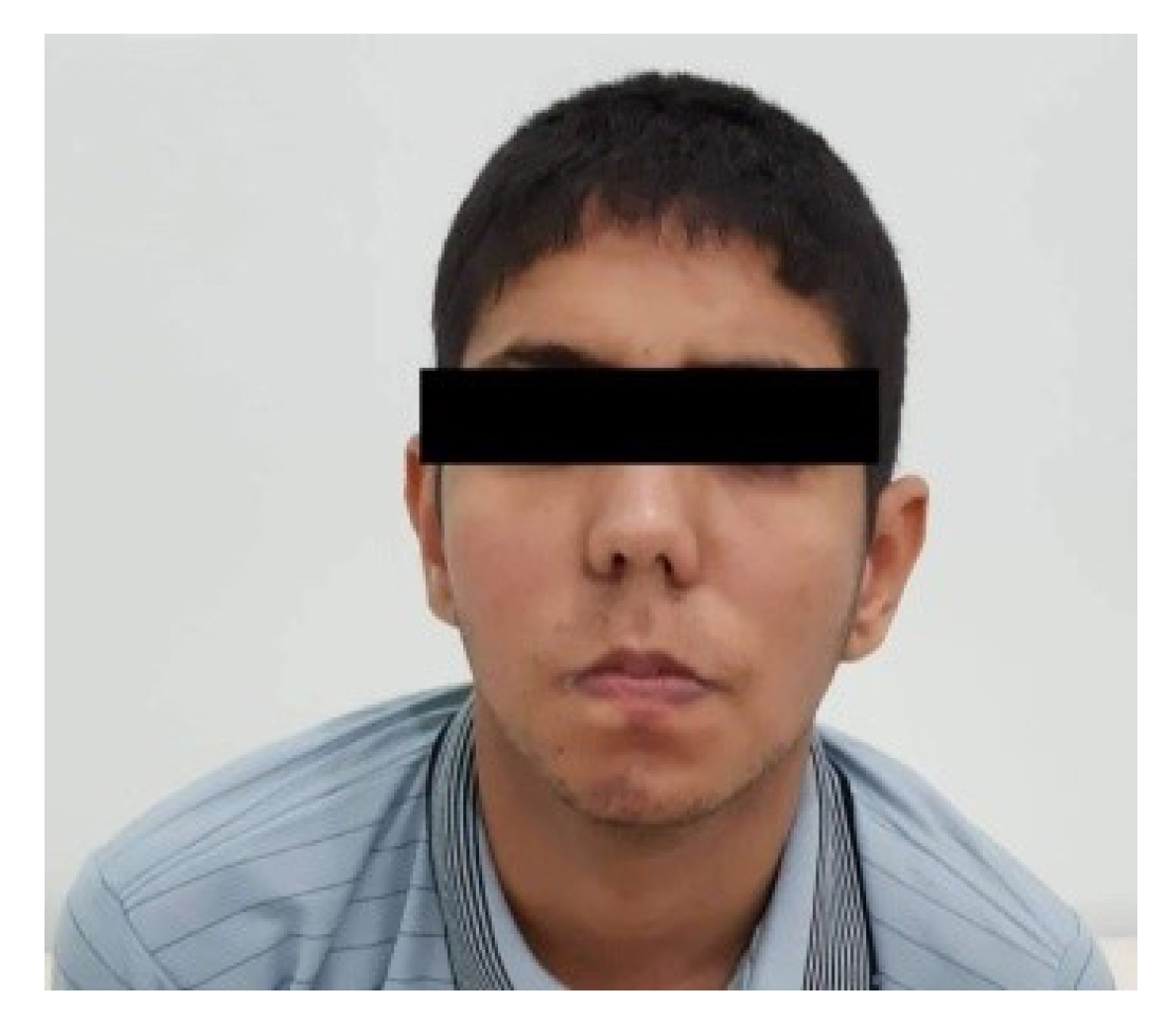
When the patient was 16 years old, a clinical examination revealed a dysmorphic syndrome characterized by microcephaly, a narrow face, ptosis, a long philtrum, and a downturned mouth (Fig. 1). Upon neurological examination, distal amyotrophy was observed, primarily affecting the thenar, hypothenar, and interosseous muscles (Fig. 2). The patient also exhibited a proximodistal motor deficit in all four limbs, with a distal predominance and pyramidal signs characterized by diffuse, vivid, and polykinetic osteotendinous reflexes. Electromyography indicated a chronic, distally predominant axonal motor neuropathy.
The patient’s weight was 30 kg, and his height was 148 cm, which fell substantially below the target height of 169 cm (standard deviation score [SDS], -3.5). His bone age was determined to be 12 years and 6 months. The patient displayed no signs of pubertal development, and genital examination revealed a bilateral testicular volume of 4 mL and a penis length of 6 cm.
Hormonal evaluation revealed normal somatotropic function. The patient’s prolactin level and thyroid function also fell within normal ranges, while assessment of the hypothalamic-pituitary-gonadal axis indicated hypogonadotropic hypogonadism. Pituitary magnetic resonance imaging (MRI) showed no abnormalities. The patient continues to receive corticosteroid replacement therapy along with fludrocortisone. He now stands at a height of 160 cm (SDS, −2.4) and exhibits normal adult genitalia, suggesting that he experienced functional hypogonadotropic hypogonadism.
A second patient, the child of a consanguineous couple, has been living with Addison disease since she was 5 years old. At the age of 23 years, she was referred to our endocrinology department. Physical examination revealed hyperpigmentation, fatigue, and weight loss. The patient also reported severe difficulty swallowing and alacrima. Consequently, she was directed to the ophthalmology clinic, where a Schirmer test was positive. Radiological barium esophageal transit and manometry studies revealed achalasia, which necessitated balloon dilatation of the esophagus. Based on these findings, a clinical diagnosis of Allgrove syndrome was established. Neurological examination disclosed a pyramidal syndrome affecting all four limbs. However, cerebral MRI revealed no abnormalities. Genetic testing identified a homozygous mutation in the AAAS gene (c.580 C>T [p.Arg194] in exon 7). The patient is now prescribed 25 mg of hydrocortisone along with artificial tears to manage her symptoms.
The third patient is a 25-year-old man was born to consanguineous parents and has a family history of hypogonadotropic hypogonadism, as one of his sisters is affected. His medical history dates back to the age of 2 years. He presented with a micropenis associated with bilateral cryptorchidism, but without anosmia. Subsequently, he was diagnosed with hypogonadotropic hypogonadism. At the age of 19 years, the patient underwent orchidopexy and began hormone replacement therapy. He also exhibited alacrima, for which he was prescribed artificial tears. At the age of 22 years, the patient developed primary AI, as evidenced by low serum cortisol levels and an elevated ACTH level of 90 pg/mL. In recent years, he reported experiencing dysphagia, which was attributed to achalasia. Given these symptoms, Allgrove syndrome was strongly suspected, prompting genetic testing. Molecular analysis identified a predominant splice donor mutation in a homozygous state (IVS14+1G→A) in the AAAS gene, confirming the diagnosis of Allgrove syndrome in this patient. Neurological examination revealed distal amyotrophy, primarily affecting the thenar, hypothenar, and interosseous muscles, as well as proximodistal motor deficiency in all four limbs, with the most pronounced effects distally. Additionally, a quadripyramidal syndrome was noted, characterized by diffuse, brisk, polykinetic osteotendinous reflexes.
All clinical and genetic characteristics of the three cases are summarized in Table 1.
Allgrove syndrome is an autosomal recessive disorder characterized by clinical and genetic heterogeneity. This condition is associated with the AAAS gene, which encodes the ALADIN protein. Mutations in this protein can result in its mislocalization to the cytoplasm, preventing it from performing its key functions [6]. The primary consequence of this mislocalization is the reduced nuclear import of DNA ligase I, aprataxin, and ferritin heavy chain [4]. These molecules are crucial for cell protection and nuclear repair, safeguarding against cellular damage caused by oxidative stress, ultraviolet radiation, and iron overload. Impaired import of these protective molecules leads to an increase in intracellular free radicals, ultimately resulting in cell death [7].
Triple A syndrome primarily affects the lacrimal glands, esophagus, and adrenal glands. The histological affinity of Allgrove syndrome remains to be determined. However, it has been shown that nucleoporin composition is not uniform throughout the body, which may partially explain the variation in tissue involvement observed in patients with this condition [6].
The classic clinical triad for this syndrome comprises alacrimia, achalasia, and Addison disease. The chronology of these manifestations varies. In the present report, patients 1 and 2 were diagnosed with AI in childhood, at the respective ages of 3 and 5 years. For patient 3, the onset of AI occurred later, in adulthood. Some authors suggest that achalasia and alacrimia may precede the onset of AI [8].
The most consistent clinical manifestation of Allgrove syndrome is alacrimia, which has a prevalence of up to 90% among affected individuals [4,9]. Parents often notice this sign from birth, although it is seldom the primary reason for seeking medical consultation; this similarly held true for our patients.
Achalasia is the second major indicator of triple A syndrome and the most common reason for medical consultation, occurring in 75% of cases [10]. The age of symptom onset can range from 6 months to 16 years [11,12]. Patients typically present with repeated vomiting, dysphagia, and bronchopulmonary complications. The prevailing pathogenic hypothesis for alacrimia and gastric achalasia involves the degeneration of cholinergic neurons within the autonomic nervous system, which leads to a progressive decline in cholinergic function [9,12,13]. Diagnosis is primarily made through esophageal manometry, although gastro-duodenal transit studies can also provide valuable information [14].
AI has a variable age of onset. Clinical signs typically emerge within the first decade of life, although they can occasionally appear before the age of 1 year. Preservation of cortisol secretion in the third decade has also been reported [2,4,15]. Symptoms that prompt a diagnosis include frequent episodes of hypoglycemia, severe fatigue, weight loss, and notably, hyperpigmentation [8,16]. The patients in the present report developed neurological manifestations during follow-up. Gazarian et al. [13] introduced the term “4A syndrome” to describe the characteristic triad of Allgrove syndrome when associated with neurological abnormalities. While some of these signs may be attributed in part to complications of hypoglycemia, the primary explanation is neuronal degeneration resulting from oxidative stress [5].
Central nervous system involvement can lead to progressively worsening intellectual disability, parkinsonism or pyramidal syndrome, epilepsy, and neurosensory disorders [17]. Cranial nerve damage may manifest as a nasal voice, lowered palate veil, or tongue fasciculations. Less common conditions such as amyotrophic lateral sclerosis [18], distal motor neuropathy, muscle hypotonia, progressive distal amyotrophy, brisk reflexes, and deep sensory disorders have also been reported [4]. Nerve conduction velocity tests typically reveal axonal motor neuropathy [17]. Muscle biopsy findings can indicate neurogenic degeneration, a non-specific myopathic process, or mixed pathology. Neurological symptoms generally emerge in the second decade of life or during adulthood, but occasionally present in childhood [4]. In our study, the onset of these symptoms occurred at ages 21, 27, and 22 years for patients 1, 2, and 3, respectively. The late presentation of the disease and atypical neurological involvement initially leads to misdiagnosis in some instances [17]. Furthermore, steroid supplementation appears to have no effect on the development or progression of neurological symptoms [4]. However, personalized physiotherapy sessions may improve endurance and balance in patients experiencing neuromuscular symptoms.
Intriguingly, in addition to the classic triad of Allgrove syndrome, patients 1 and 3 exhibited endocrine disorders. Specifically, these involved growth and pubertal delay, which were attributed to Addison disease. These patients demonstrated poor compliance with substitution therapy and were repeatedly hospitalized for acute AI resulting in dysfunction of the hypothalamic-pituitary axis.
Mutations were found throughout the AAAS gene, suggesting the absence of a hotspot [11]. Molecular analysis identified the splice site c.1331+1G>A mutation in the homozygous state for both patients 1 and 3. This major mutation (G>A) is a substitution at the first position of intron 14. It abolishes the splice donor site, leads to the production of abnormal transcripts of variable size, and triggers premature termination of translation [19]. In contrast, patient 2 exhibited a nonsense mutation at exon 7 (a C>T substitution at nucleotide position 580), which resulted in the substitution of arginine at amino acid position 194 with a stop codon (p. Arg194stop) [20]. AI and alacrima are the two primary signs in patients with the splice site c.1331+1G>A mutation [9]. In nearly all cases, no genotype-phenotype correlation is noted, and substantial intrafamilial and interfamilial heterogeneity is observed. That is, patients with identical mutations—even those within the same family—may exhibit variable clinical presentations. Therefore, phenotypic expression must be influenced by currently unknown modifying factors [10]. The geographic clustering of apparently unrelated patients with the same mutations has led several authors to propose regional founder effect for some recurrent mutations, a hypothesis supported by haplotype analysis [11]. This particular mutation is believed to be inherited from a common ancestor and is widespread in North African populations. The splice site mutation c.1331+1G>A has also been observed in families from Spain, Mediterranean populations, Puerto Rico, Mexico, and Iran. Haplotype analysis in several studies has suggested that this mutation originated in the North African population approximately 1,000 to 1,175 years ago.
We report three cases of Allgrove syndrome accompanied by neurological manifestations. Neurological disorders are frequently encountered in this context, but they can be easily overlooked. Therefore, a thorough neurological examination should be a routine component of the evaluation for patients with this syndrome. Additionally, genetic counseling should be made available to all patients.