The Genetic Facets of Dravet Syndrome: Recent Insights
Article information

Abstract
Dravet syndrome (DS), previously known as severe myoclonic epilepsy of infancy, is a severe epileptic syndrome affecting children, with an incidence of 1/22,000 to 1/49,900 live births annually. Characterized by resistant and prolonged seizures, it often leads to intellectual impairment, with males being twice as susceptible as females. Its clinical features include recurrent seizures triggered by fever initially, but later occurring spontaneously, developmental delays, behavioral issues, and movement disorders. Sodium voltage-gated channel alpha subunit 1 (SCN1A) mutations, observed in about 90% of cases, are usually de novo, while mutations in other genes, such as protocadherin 19 (PCDH19), gamma-aminobutyric acid type A receptor subunit gamma 2 (GABRG2), and sodium voltage-gated channel alpha subunit 2 (SCN2A), can also contribute to the condition. Next-generation sequencing aids in identifying these genetic abnormalities. First-line treatments include anticonvulsant drugs such as valproate, clobazam, stiripentol, topiramate, and bromide. Second-line treatments for drug-resistant DS include stiripentol, fenfluramine, and cannabidiol. This literature review provides a comprehensive update on the genetic underpinnings of DS, highlighting SCN1A's predominant role and the emerging significance of other genes. Moreover, it emphasizes novel therapeutic approaches for drug-resistant forms, showcasing the efficacy of newer drugs such as stiripentol, fenfluramine, and cannabidiol. This synthesis contributes to our understanding of the genetic landscape of DS and informs clinicians about evolving treatment strategies for enhanced patient care.
Introduction
Dravet syndrome (DS) was initially described by Charlotte Dravet in 1978 as a new epilepsy syndrome, called severe myoclonic epilepsy of infancy (SMEI) [1]. DS has been recently defined as a developmental and epileptic encephalopathy. It is a type of epilepsy that occurs in early life, with recurrent febrile and afebrile convulsive seizures, which are often prolonged.
Recent reviews have highlighted the intricate electroclinical profile of DS and the nuanced relationship between sodium voltage-gated channel alpha subunit 1 (SCN1A) mutations and clinical manifestations, extending beyond seizures to comorbidities. Notably, besides SCN1A, several genes are implicated in DS-like phenotypes, emphasizing the expanding genetic landscape underlying this complex epileptic condition. This genetic diversity challenges a purely syndromic classification, urging a nuanced approach for comprehensive understanding and treatment strategies. Guidelines have proposed standardized treatments, encompassing established antiseizure medications and emerging options such as cannabidiol (CBD) and fenfluramine, with notable impacts on quality of life and cognition [2,3].
In this review, we discuss the clinical presentation of DS, its genetic causes, and advances in its treatment. Overall, the review provides a thorough overview of the genetic basis of DS and the potential for gene therapies to treat this devastating disorder.
Clinical Presentation
Typically, seizures manifest in previously healthy children within the first year of life and are characterized by prolonged, febrile, and afebrile hemiclonic or generalized clonic seizures. These seizures may be triggered by factors such as vaccinations or hyperthermia. In the following months, affected individuals experience recurring seizures, both febrile and afebrile, often affecting different sides of the body. Between the ages of 1 and 4, additional seizure types emerge, including myoclonic seizures, atypical absences, focal seizures, and generalized tonic-clonic seizures. Focal seizures can manifest with autonomic features such as pallor, cyanosis, and drooling, and may progress to focal motor or bilateral convulsive seizures. Tonic seizures are less commonly observed in DS. In some individuals, myoclonic seizures may not develop, and other seizure types, particularly focal or multifocal seizures, become the predominant presentation.
Some DS patients may also have photosensitive epilepsy. Along with epileptic seizures, DS and other comorbidities that can coexist include ataxia, premature death, language and motor development delay, cognitive impairment, sleep disorders, autism spectrum disorder, and sudden unexpected death in epilepsy [4].
In a retrospective survey of 138 Chinese children with DS with mutations in the SCN1A gene, the onset of the first seizure occurred before the age of 7 months in 77% of cases. In the same study, 72% of children had febrile convulsions lasting more than 15 minutes, and 67% had two or more febrile seizures over a 24-hour period. The convulsions were hemiclonic, with fever present in 80% of cases [5].
Regarding the progression of epilepsy in individuals with DS, there is a tendency for seizures to become less frequent and severe during adolescence and adulthood. While fever sensitivity persists, its impact tends to be reduced. Myoclonic seizures, atypical absence seizures, and focal seizures with altered awareness are less common in adulthood [6]. The most prevalent seizure type in adults with DS is generalized tonic-clonic seizures, which may have a focal onset and predominantly occur during sleep. Additionally, patients may experience bilateral or asymmetrical posturing, followed by tonic vibratory or clonic movements. However, there is a limited number of studies providing detailed information on the electroclinical features of seizure types in DS during adolescence and adulthood.
Electroencephalography (EEG) in patients with DS exhibits distinctive features that play a crucial role in understanding and diagnosing this complex epileptic condition. Several studies have explored the details of EEG in patients with DS; for instance, a meta-analysis synthesized EEG data from 155 patients with DS to unveil how EEG characteristics evolve with age [7]. The results of this meta-analysis highlighted significant trends in the progression of brain electrical signals in this syndrome.
EEG is typically normal at the time of first presentation, but various abnormalities may be seen later. The most common EEG abnormalities observed in patients with DS are generalized or focal slowing, generalized spike-wave, and focal or multifocal spikes or spike-wave discharges with normal interictal EEG. Later, interictal EEG characteristics include generalized, focal, and multifocal abnormalities, as well as strong photosensitivity in up to 40% of patients [2]. These abnormalities can be useful in differentiating DS from other entities, such as genetic epilepsy with febrile seizures plus (GEFS+) [7-10].
The age of the patient has a significant impact on temporal EEG changes in DS. In the above meta-analysis of EEG patterns as a function of age in 155 patients, EEG was abnormal in 43% of patients aged 0 to 12 months, but this rose to 90% for the 1- to 2-year-old group, and remained at approximately the same level for the remainder of the age groups. The earliest reported ages of generalized epileptiform discharges, focal epileptiform discharges, diffuse background slowing, and focal slowing were 6, 4, 4, and 4 months, respectively.
Genetics of Dravet Syndrome
DS is primarily caused by mutations in the SCN1A gene. Over 90% of cases are due to a de novo mutation in one allele of this gene, which encodes the α-subunit of the voltage-gated ion channel NaV1.1 [11].
It is important to note that our understanding of the genetic landscape of DS is still evolving, and ongoing research may uncover additional genes associated with the condition. Additionally, while mutations in these genes are linked to DS, the precise mechanisms through which they contribute to the disorder's pathophysiology are still being investigated.
This section will delve into the pathophysiology of DS, involving not only the SCN1A gene but also other genes associated with the syndrome's manifestation. However, recent studies have identified other genes that are also involved in DS and related phenotypes [4].
1. SCN1A variants
Voltage-gated sodium channels (VGSCs) play a pivotal role in ensuring the proper functioning of the nervous system [12], particularly in the initiation and propagation of action potentials. To date, nine distinct sodium channel subunits have been identified and validated, denoted as Nav1.1 to Nav1.9. These channels consist of four separate but homologous domains labeled DI to DIV, each encompassing six transmembrane segments designated as S1 through S6 (Fig. 1) [13]. SCN1A is positioned on chromosome 2 at locus 24.3 (2q24.3), and the SCN1A gene spans approximately 85 kb and is composed of 26 coding exons [3].
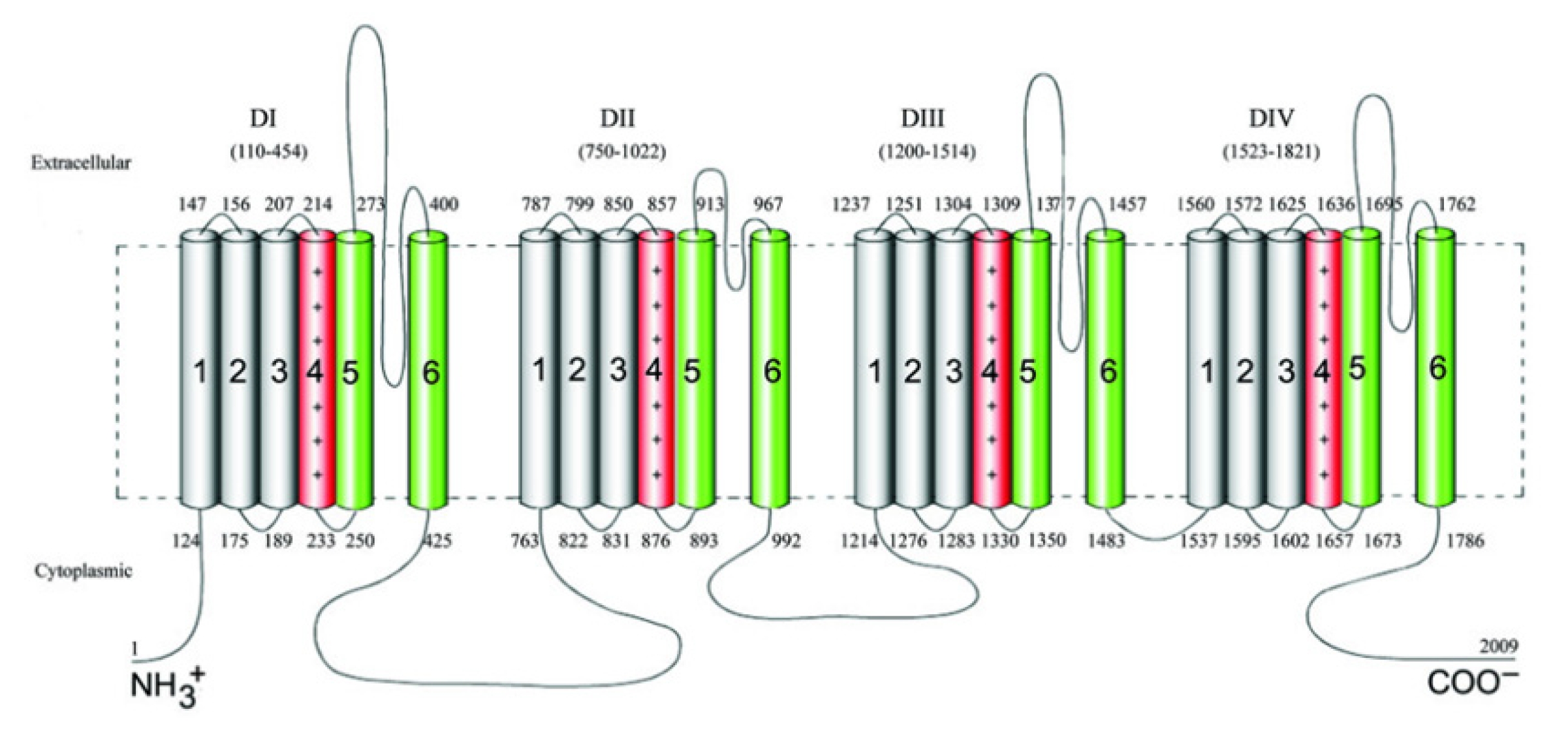
The VGSC a1 subunit (NaV1.1), which functions as the only pore-forming subunit, is encoded by the SCN1A gene [14]. This subunit holds significant prominence as a major contributor to epileptic conditions and serves as the primary pathogenic gene associated with DS.
Not all SCN1A mutations result in DS, and SCN1A is linked to other diseases. SCN1A is one of the most often mutated genes that cause GEFS+. In fact, GEFS+ and DS are two distinct forms of epilepsy that are brought on by SCN1A mutations. There have been reports of additional epilepsies linked to SCN1A, including Doose syndrome, epilepsy of infancy with migrating focal seizures, West syndrome, Lennox-Gastaut syndrome, and early infantile epileptic encephalopathy (EIEE) with more severe mutations than occur in DS. SCN1A is also linked to a few non-epileptic conditions, such as autism spectrum disorder, arthrogryposis multiplex congenita, and hemiplegic migraine [15].
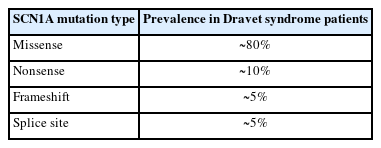
Around 1,800 pathogenic variations in the SCN1A gene have been found in people with DS and other types of epilepsy [16]. Missense variants appear in one-third to 80% of DS cases (Fig. 2), whereas truncating variations, including nonsense, frameshift, and splice site variants, are seen in over 20% of cases (Table 1, Fig. 3) [12,17-20]. Deletions of the whole gene, exons, and promotor regions are some other variations [17,21,22]. It is interesting to note that a recent study found numerous variations in SCN1A's intron 20 that promote the inclusion of a poison exon between exon 20 and exon 21, resulting in truncation and a smaller full-length functional channel [23]. The poison exon is a regulatory exon that triggers the degradation of mRNA when included in the transcript. In a mouse model, the poison exon was inappropriately included due to a non-coding variant, leading to the degradation of SCN1A mRNA [24].
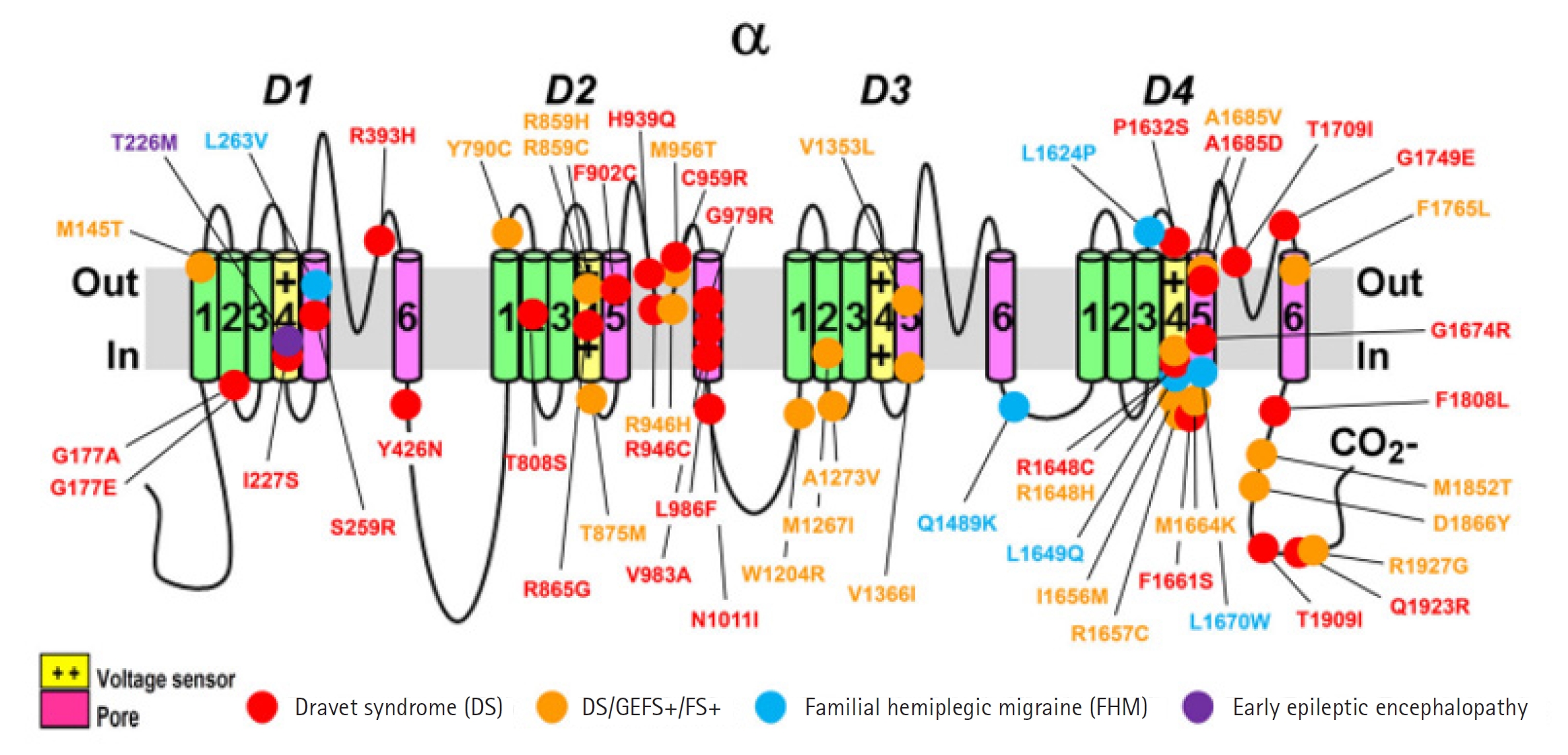
The types of SCN1A mutations associated with DS and their prevalence in patients.
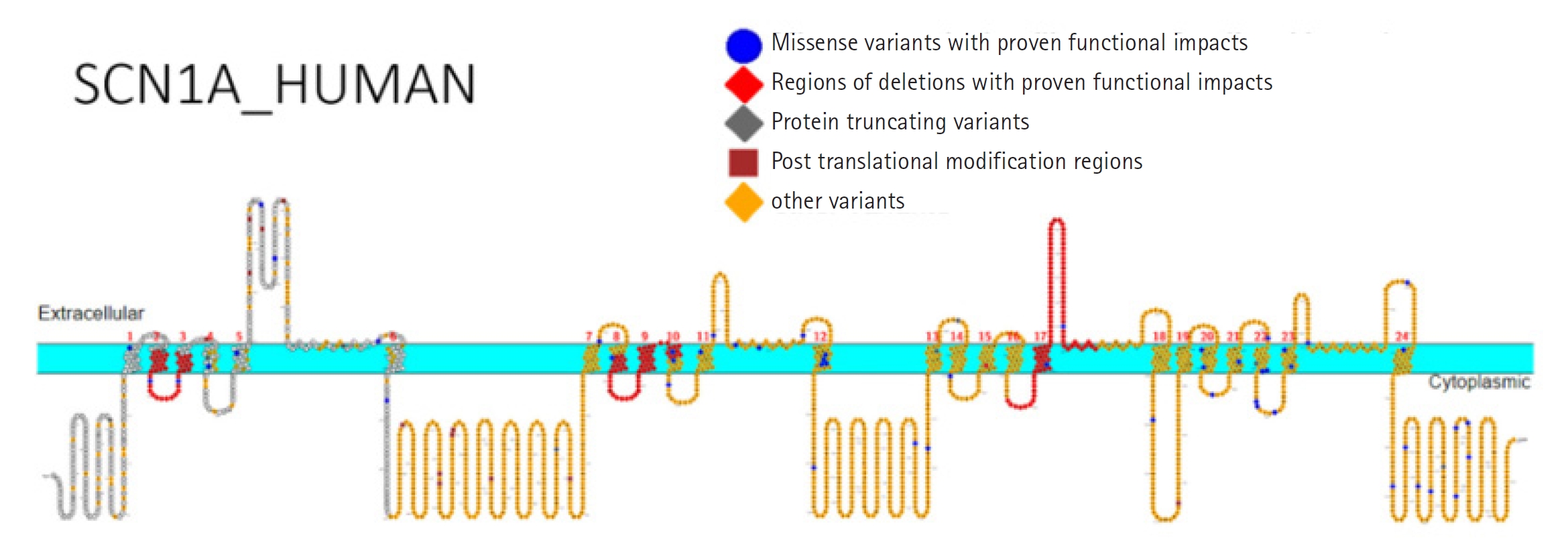
Primary structure of the sodium voltage-gated channel alpha subunit 1 (SCN1A) protein showing regions with functionally validated epilepsy-variants from literature. Protein-truncating variant region (black), regions of deletions with proven functional impacts (red), regions of missense variants with proven functional impacts (blue), regions of post-translational modifications (brown), and other variants (orange). Created using Protter [19]. Visualization link: https://bit.ly/2NWfncp [20].
Missense mutations, which result in a change in a single amino acid in the SCN1A protein, are the most common type of mutation and are estimated to account for approximately 85% of cases. Nonsense mutations, frameshift mutations, and splice site mutations are less common but still contribute to the development of DS. This information is important for understanding the genetic basis of the disease and may have implications for diagnosis and treatment.
2. Physiopathology of SCN1A in DS
It is clear that DS predominantly arises from diminished NaV1.1 functionality due to haploinsufficiency. Mutations in the SCN1A gene disrupt the balance between neuronal excitation and inhibition in the brain, leading to epileptic seizures and other symptoms of DS [25]. Electrophysiological scrutiny of DS-linked missense variants underscores their tendency to provoke loss-of-function alterations, often rendering channels nonfunctional. Nevertheless, select variants exhibit gain-of-function outcomes, accentuating the intricate impact of NaV1.1 variants in DS pathophysiology [26-28].
Studies using mouse models have shed light on how the dysfunction of sodium channels, particularly NaV1.1, results in epilepsy [29]. While pyramidal neurons in the cerebral neocortex and hippocampal formation are less impacted, the deterioration in function is characterized by decreased sodium current density and action potential firing in GABAergic interneurons [30,31]. Researchers have demonstrated that the basic characteristics of DS, such as temperature-induced seizures, spontaneous seizures, early mortality, and cognitive and social deficits, may be replicated in mice by specifically knocking out Scn1a in forebrain GABAergic interneurons [32,33]. Additionally, parvalbumin-positive interneurons, which are essential for controlling the firing of pyramidal neurons and inhibitory function in the brain, express NaV1.1 mostly in the beginning segments of the axon [30]. These results imply that the main mechanism underlying DS is the reduced cerebral inhibition brought on by NaV1.1 haploinsufficiency. Human cell studies utilizing induced pluripotent stem cells have further supported this concept [34-36].
Similar to this, studies on the pathophysiology of SCN1A mutations in DS have revealed that the loss of this ion channel's functionality causes neuronal hyperexcitability and decreased inhibitory signaling, which may aid in the onset of epileptic episodes. Studies on mouse models of DS have also provided insights into potential therapeutic approaches, such as targeting specific neurotransmitter systems and modulating ion channel function, to reduce the symptoms of this devastating disorder [37].
Voskobiynyk et al. [24] (2021) used Clusters of Regularly Interspaced Palindromic Repeats (CRISPR)/Cas9 genome editing to introduce a non-coding variant into wild-type mice, which resulted in aberrant splicing of Scn1a mRNA and a decrease in SCN1A protein expression. The mice exhibited increased susceptibility to seizures and premature death, similar to the phenotype observed in DS patients with SCN1A mutations [24].
3. Other genes associated with DS
In a literature review, Ding et al. [4] identified several genes that are associated with DS and similar phenotypes, including SCN2A, sodium voltage-gated channel alpha subunit 8 (SCN8A), protocadherin 19 (PCDH19), and gamma-aminobutyric acid type A receptor subunit alpha1 (GABRA1). Additionally, other genes have been implicated in similar phenotypes, such as syntaxin-binding protein 1 (STXBP1), potassium voltage-gated channel subfamily A member 2 (KCNA2), potassium voltage-gated channel subfamily Q member 2 (KCNQ2), KCNQ3, solute carrier family 2 member 1 (SLC2A1), and glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A). These genes encode proteins involved in neuronal excitability, synaptic transmission, and metabolism (Table 2) [15,16,38-62].

Several genes linked to Dravet syndrome
Despite these advances, the pathogenesis of DS is not fully understood. The role of each gene in the development and progression of the syndrome remains to be fully elucidated, and there may be additional genes that contribute to the disorder. Further research is needed to better understand the underlying mechanisms of DS and related phenotypes, which could inform the development of more effective treatments for this debilitating disorder [4].
1) SCN2A
The SCN2A gene is located on chromosome 2q24.3 and encodes the alpha subunit of the VGSC Nav1.2. This channel is primarily expressed in the central nervous system and plays a critical role in the initiation and propagation of action potentials in neurons. Missense mutations in SCN2A result in altered channel function and excitability of neurons, leading to seizures and other neurological symptoms. SCN2A mutations have been associated with DS and other forms, including infantile spasms, focal epilepsy, and GEFS+.
In a study including 59 Japanese patients who had been diagnosed with DS (SMEI, n=33; and borderline SMEI, n=26). The study found that three novel missense mutations of SCN2A were identified in three (5.1%) of 59 patients but were not found in the 96 healthy volunteers. The mutations affected highly conserved amino acids in many species and were located in different domains of the SCN2A protein. The study suggests that missense mutations of SCN2A can be a cause of DS, expanding the sodium channel mutation spectrum [38].
2) SCN8A
The SCN8A gene is located on chromosome 12q13.13 and encodes the Nav1.6 VGSC. Gain-of-function mutations in SCN8A have been associated with DS and other severe EIEE. Unlike Nav1.1, the distribution pattern of this sodium channel differs, being specifically expressed on the dendritic spines of cortical pyramidal neurons, which are the primary excitatory cells in the neocortex. In contrast to the inhibitory role of Nav1.1, Nav1.6 primarily functions as an excitatory channel [2]. Loss-of-function mutations have been reported, but their association with epilepsy is less clear [39].
Through clinical exome sequencing, a recent investigation discovered three previously unreported de novo mutations within the SCN8A gene [39,63]. The mutations resulted in varying clinical features, including epilepsy and movement disorders, and varying degrees of developmental delay. Electrophysiological analysis revealed distinct effects of the mutations on channel activity, with one causing gain-of-function, one causing apparent loss-of-function, and one having normal channel activity. Research suggests that gain-of-function mutations of SCN8A result in severe EIEE, while partial loss-of-function mutations result in later-onset seizures and severe developmental delay [39,64]. The data demonstrate the importance of functional testing in establishing the pathogenicity of de novo mutations.
Pathogenic heterozygous variants have been extensively documented as causative factors for epileptic encephalopathy, with the age of onset varying from infancy to 18 months. Nonetheless, the fundamental phenotype associated with SCN8A differs notably from that of DS. Notably, while epileptic spasms are observed in a minority of SCN8A encephalopathy cases, they do not characterize DS. Furthermore, certain DS attributes, such as fever sensitivity and myoclonic seizures, are frequently absent in SCN8A encephalopathy cases. The range of EEG findings is diverse, encompassing initial hypsarrhythmia to prolonged normal patterns. Nonetheless, many patients display background slowing, as well as focal or multifocal sharp waves or spikes.
It is important to emphasize that some patients exhibited favorable responses to sodium channel blockers such as carbamazepine, oxcarbazepine, or phenytoin, which are typically avoided in DS due to their potential to exacerbate seizures. While some individuals in the original SCN8A cohort were initially tentatively diagnosed with DS, it is now widely accepted as a distinct condition [2].
3) SCN9A
The sodium voltage-gated channel alpha subunit 9 (SCN9A) gene is located on chromosome 2q24.3 and encodes the alpha subunit of the Nav1.7 sodium channel. Mutations in the SCN9A gene have been linked to several conditions, including DS, febrile seizures, and congenital insensitivity to pain.
The implications of the SCN9A gene in DS have been examined in multiple studies. One study identified mutations linked to epileptic encephalopathy, illustrating a gain-of-function phenotype within sodium channels produced by SCN9A [40]. This heightened neuronal excitability may contribute to the characteristic features of DS. Moreover, the researchers observed that these mutations displayed distinct sensitivity to the antiepileptic drug (AED) oxcarbazepine, suggesting potential implications for treatment strategies. Another study highlighted SCN9A mutations in patients with febrile seizures and DS, hinting at SCN9A's potential role as a genetic modifier impacting the severity of DS symptoms. Collectively, these studies revealed that SCN9A mutations induce neuronal hyperexcitability through a gain-of-function phenotype, solidifying the significance of this gene as a pivotal contributor to the development of DS [65].
In a study [66], it was observed that SCN9A gene mutations were relatively rare in patients with congenital insensitivity to pain and erythromelalgia, but the mutations that were found were associated with more severe symptoms. This study reported a de novo SCN9A mutation in a patient with DS who did not have a mutation in SCN1A. The mutation was predicted to result in a gain-of-function effect on Nav1.7, leading to increased neuronal excitability. The patient had a history of prolonged seizures, developmental delay, and cognitive impairment, which are characteristic features of DS.
Mulley et al. [67] (2013) performed a genome-wide association study to identify genes that were associated with DS. They found that the SCN9A gene was the most significantly associated gene and has been implicated in the development of DS. However, some DS cases without SCN1A mutations had predicted SCN9A susceptibility variants, which may be attributed to the complex inheritance patterns for these unexplained cases of DS. Compared with controls, DS cases were significantly enriched for rare SCN9A genetic variants. However, none of the multiplex febrile seizure or GEFS+ families could be explained by highly penetrant SCN9A mutations. Therefore, SCN9A may play a role in the development of DS, particularly in cases without SCN1A mutations, and it may also contribute to the severity of symptoms in both febrile seizures and DS. However, further research is needed to fully understand its involvement in this disorder.
4) SCN1B
The SCN1B gene is located on chromosome 19q13.1 and encodes the sodium channel beta-1 subunit. Mutations in the SCN1B gene have been associated with various forms of epilepsy, including DS and GEFS+.
A previous study investigated the contribution of sodium channel 1 subunits in mice to the regulation of neuronal excitability and nodal architecture [48]. The researchers used mice lacking either the alpha or beta subunit of the sodium channel 1 gene and performed a variety of experiments to assess the effects of these subunits on sodium channel expression, nodal architecture, and neuronal excitability. The study found that mice lacking the beta subunit of the sodium channel 1 gene had reduced expression of sodium channels and altered nodal architecture in the optic nerve, which led to decreased conduction velocity and increased threshold for action potential initiation. These mice also exhibited reduced excitability in cortical neurons and decreased susceptibility to seizures induced by the convulsant drug pentylenetetrazol.
There have been two reported examples of pathogenic homozygous SCN1B mutations causing DS. The first was a child born to consanguineous parents who started having febrile seizures at 3 months old. He thereafter had severe hypotonia, developmental regression, tonic-clonic convulsions, and severe myoclonus, all of which were frequently accompanied by fevers. Rolandic discharges were seen on the EEG. He had seizures that were resistant to medication, including clobazam, and at the age of 13 months, aspiration pneumonia claimed his life. This person's SCN1B intracellular expression was normal, but there was no cell surface expression, making them a functional null variation [68]. According to a different research, a person's development was normal up to the age of 6 months, at which point fever-sensitive seizures of various sorts, including myoclonic ones, as well as developmental standstill at the time of epilepsy's beginning, global developmental delay, ataxia, and persistent uncontrollable epilepsy began. Interictal EEG was originally normal but started to show multifocal spikes and slow waves, either singly or in bursts, around the age of 1 year [69].
Following these results, 54 DS patients were examined for SCN1B variations but were negative for SCN1A variants; no SCN1B variants were discovered [70]. SCN1B heterozygous, possibly pathogenic mutations with varied penetrance have been identified in a number of families with GEFS+ [71].
5) PCDH19
The PCDH19 gene is situated on the X chromosome at the Xq22.1 locus. It was initially documented in 1971, albeit without a known genetic etiology at the time [41]. Subsequently, in 1997, it was discerned that epilepsy and mental retardation limited to females (EFMR) might not align with other female-restricted disorders marked by male lethality, but instead, could exhibit male sparing [72]. This premise was substantiated by the first series of EFMR cases involving pathogenic variants linked to a DS-like phenotype. Among the 11 female cases, the series included one instance of male mosaicism, thus augmenting the body of evidence [73].
A series of studies has contributed to the growing recognition of the relevance of the PCDH19 gene in the genesis of DS and related conditions. A comparative analysis revealed that fever sensitivity, a defining characteristic of both PCDH19-related epilepsy and DS, exhibited a significantly higher prevalence in PCDH19-related cases, with approximately 90% of individuals demonstrating this trait compared to around 20% in DS cases [74]. Moreover, genetic screening of a cohort of Chinese children with DS identified a notable co-occurrence of mutations in both SCN1A and PCDH19 genes, accounting for approximately 15% of the patients studied [75]. In the context of sporadic infantile epileptic encephalopathy attributed to PCDH19 mutations, a distinct sex bias was observed, with over 90% of patients being female, offering an important sex-specific perspective to the genetic underpinnings of PCDH19-related phenotypes [73].
Further insights into the genetic landscape of DS were highlighted in a comprehensive literature review, where genetic factors beyond SCN1A were implicated in approximately 10% of cases, revealing the expanding role of various genes, including PCDH19, in the clinical spectrum of the disorder [2]. The significance of PCDH19 mutations was reinforced through a systematic review, demonstrating their presence in approximately 30% of patients with DS-like phenotypes, further solidifying their relevance in the complex genetic architecture of these early-onset epilepsies [76].
Furthermore, investigations into the genetic basis of sporadic infantile epileptic encephalopathy caused by PCDH19 mutations have revealed insights into the distinct sex-specific expression of this condition. This sex-biased presentation, predominantly affecting females, adds a unique dimension to the complexity of PCDH19-related epilepsies. A comprehensive review of SCN1A and PCDH19 variants further elucidated the genetic heterogeneity underlying DS and its phenotypic mimics [77]. This compilation not only emphasized the necessity of precise genetic characterization for accurate diagnosis but also underscored the potential for tailored therapeutic strategies. Additionally, therapeutic avenues were explored, indicating the immediate suppression of seizure clusters through corticosteroid administration in PCDH19 female epilepsy [77]. This intervention's efficacy sheds light on acute management strategies for controlling seizure episodes.
6) GABRA1
The GABRA1 gene is located on the 5q34 chromosome and has been implicated in various forms of epilepsy, including DS. In 2014, a comprehensive methodology was utilized by researchers to identify genetic factors associated with DS. Employing whole-exome sequencing, the study included a cohort of affected individuals, including both sporadic and familial cases. Within the scope of identified genetic determinants, mutations in the GABRA1 gene emerged as a significant revelation, contributing to a substantial portion of cases. Remarkably, these mutations were detected in approximately 8% of the examined cohort, underscoring the pivotal role of GABRA1 in the pathogenesis of DS [42].
A study conducted an in-depth phenotypic analysis on individuals harboring GABRA1 mutations, extending beyond the confines of DS. The investigation encompassed a diverse spectrum of epilepsy phenotypes, aiming to clarify the broader implications of GABRA1 mutations. The findings unveiled a wide array of clinical manifestations, including varying degrees of seizure severity and associated features. Notably, approximately 15% of individuals with GABRA1 mutations exhibited clinical presentations consistent with DS, while the remaining cases displayed a range of other epilepsy phenotypes [78].
In summary, the studies conducted to establish the involvement of the GABRA1 gene in the genesis of DS have been characterized by robust genetic analyses and comprehensive clinical evaluations. The substantial percentages of affected individuals with GABRA1 mutations within various cohorts emphasize the gene's significant contribution to the pathogenesis of DS. These percentages provide quantitative insights into the prevalence and relevance of GABRA1 mutations, thereby enhancing our understanding of the intricate interplay between genetics and disease manifestation.
7) GABRG2
The gamma-aminobutyric acid type A receptor subunit gamma 2 (GABRG2) gene has a chromosomal location of 5q34. An extensive review by researchers [2] explored the genetic complexities of DS and its mimics, moving beyond the confines of SCN1A. This comprehensive analysis synthesized data from diverse studies, shedding light on a broader genetic landscape. While precise percentages were not provided, this review's importance lies in its comprehensive assessment of the diverse genetic contributors, including the GABRA1 gene in DS-related conditions.
A family with GEFS+, one of whose members had DS, has been linked to GABRG2 variations. It is noteworthy that this person also had an aunt who had a history of febrile seizures and a sister who had myoclonic-atonic epilepsy but who tested negative for GABRG2 mutations [43]. This shows that the family's epilepsy inheritance was complicated and could not be attributed simply to mutations in GABRG2, yet a causal role for the gene cannot be completely ruled out. In other families with GEFS+, inherited variations have been reported [79]. Two cases in a group of eight individuals with de novo pathogenic GABRG2 mutations and epileptic encephalopathy appeared to fit the essential diagnostic criteria for DS [80].
8) STXBP1
The STXBP1 gene, also known as MUNC18-1, is located on chromosome 9 at position 9q34.11. Coding for syntaxin-binding protein 1, it is thought to be involved in vesicle membrane fusion and, therefore, neurotransmitter release. STXBP1 epilepsy appears to be characterized by tonic seizures and/or spasms, both of which are unusual in DS [45]. There has been one report of a dramatic effect of levetiracetam antiepileptic with clonic seizures refractory to phenobarbital, pyridoxine, and phenytoin in EIEE due to a pathogenic STXBP1 variant [81].
In the pursuit of novel genetic causes, Carvill et al. [42] (2014) investigated the genetic underpinnings of DS, revealing the involvement of STXBP1 mutations. This investigation involved a systematic genetic analysis of affected individuals, leading to the identification of mutations in these genes. The findings shed light on the genetic heterogeneity of DS and emphasized the contribution of STXBP1 to the disorder's genetic architecture [42].
Furthermore, the role of STXBP1 in EIEE was established through studies [45,81-83]. These studies employed a combination of genetic analyses and clinical characterizations to delineate the genetic and clinical relevance of STXBP1 mutations. The investigations revealed the prevalence of de novo mutations in the STXBP1 gene in individuals with EIEE. The relevance of these findings is underscored by the significant proportion of affected individuals with STXBP1 mutations, highlighting the critical role of STXBP1 in the pathogenesis of this neurodevelopmental disorder.
9) HCN1
The HCN1 gene, which encodes potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 1, is found on chromosome 5 at location 5p12. The Ih current, which is involved in the spontaneous rhythmic activity of neurons, is influenced by this nonselective cation channel [47].
Six distinct missense variations in HCN1 were found after testing 39 people with fever-sensitive epileptic encephalopathy who were said to have clinical features of DS. These mutations mostly resulted in gain-of-function, according to functional testing. Age at first seizure start ranged from 4 to 13 months, and all patients experienced both febrile and afebrile seizures. Four of the six showed characteristics of autism, two had ataxia, and all six had some sort of learning difficulties. The six patients with various HCN1 missense variations all experienced atypical absence seizures in addition to a variety of other seizure types, including focal seizures in five patients, myoclonic and tonic-clonic seizures in two patients, and status epilepticus in four patients [84].
10) CHD2
CHD2 encodes chromodomain helicase DNA-binding protein 2, which modifies gene expression, probably by acting on chromatin structure [44]. It is located on chromosome 15 at location 15q26.1. An innovative study was conducted using exome resequencing in a cohort of individuals with SCN1A-negative epileptic encephalopathies. The researchers identified three heterozygous de novo mutations in the CHD2 gene. All three initially presented with febrile seizures in the second, third, or fourth year of life, which is later than is typical for DS. All went on to develop a range of seizure types, including myoclonic seizures, generalized tonic-clonic seizures, and absences. All three had normal early development, but subsequently showed mild to moderate intellectual disability and, in two cases, mild ataxia. In one case, clobazam controlled the seizures, but the other two remained intractable. The EEG showed generalized spike-wave complexes in all cases [85].
In a distinct study, a cohort of 10 individuals with established CHD2 variants and epileptic encephalopathy was meticulously examined. These children exhibited seizure onset spanning from 12 to 42 months, with an average onset age of 26 months. Notably, seven of these individuals displayed pronounced photosensitivity, a characteristic occasionally observed in DS but not frequently encountered in most of the discussed DS mimics. Myoclonic seizures were a consistent feature among all participants, and the majority experienced typical or atypical absences as well as generalized tonic-clonic seizures. Among the cohort, seven individuals exhibited varying degrees of intellectual disability, ranging from moderate to severe, and presented with drug-resistant epilepsy. Conversely, three participants displayed a slightly delayed onset, exhibited improved responses to medication, and demonstrated more favorable developmental outcomes [86]. Importantly, although some individuals in this cohort exhibited a clinical profile closely resembling DS, they were not initially classified as DS patients. This could be attributed to the availability of early genetic diagnosis, which contributed to their distinct categorization [2].
11) KCNA2
The KCNA2 gene encodes the potassium voltage-gated channel subfamily. Specifically, on the chromosomal location 1p13.3 of chromosome 1, there is a gene known as member 2 (Kv1.2) of this potassium channel subfamily. Kv1.2, a representative member of potassium channels exhibiting delayed rectification, plays a crucial role in facilitating the repolarization process in active neurons.
Six patients with epileptic encephalopathy, some of whom were first thought to have DS, had pathogenic KCNA2 mutations. There were four distinct variations altogether, of which two (four cases) resulted in a loss-of-function and two (two cases) in a gain-of-function. Despite the limited sample sizes, there seemed to be considerable phenotypic variance between the two groups. The loss-of-function individuals all attained seizure independence by maturity, which is unusual in DS, and all had onset between 8 and 17 months, mild to moderate intellectual impairment, and minimal or absent ataxia. EEG indicated acute waves that were focal or multifocal. Seizure patterns varied, with myoclonic and focal seizures with decreased awareness occurring twice as often as febrile seizures and focal motor seizures (three each). With beginning at 5 to 6 months, moderate to severe intellectual impairment and ataxia, as well as continuous generalized tonic-clonic seizures into adulthood, the two gain-of-function individuals exhibited a more severe phenotype. Generalized spike-wave discharges were seen in their EEG tests [46]. It seems that not just one, but two different novel entities may contribute to the development of KCNA2 encephalopathy.
Novel Therapeutic Approach
A recent article published in Epilepsia [87] proposed a novel strategy for predicting the efficacy of drugs for monogenic epilepsies, including DS. The authors used a systems medicine approach that combined computational modeling and experimental data to predict how specific drugs would affect the function of the mutated genes associated with the disorder (Fig. 4).
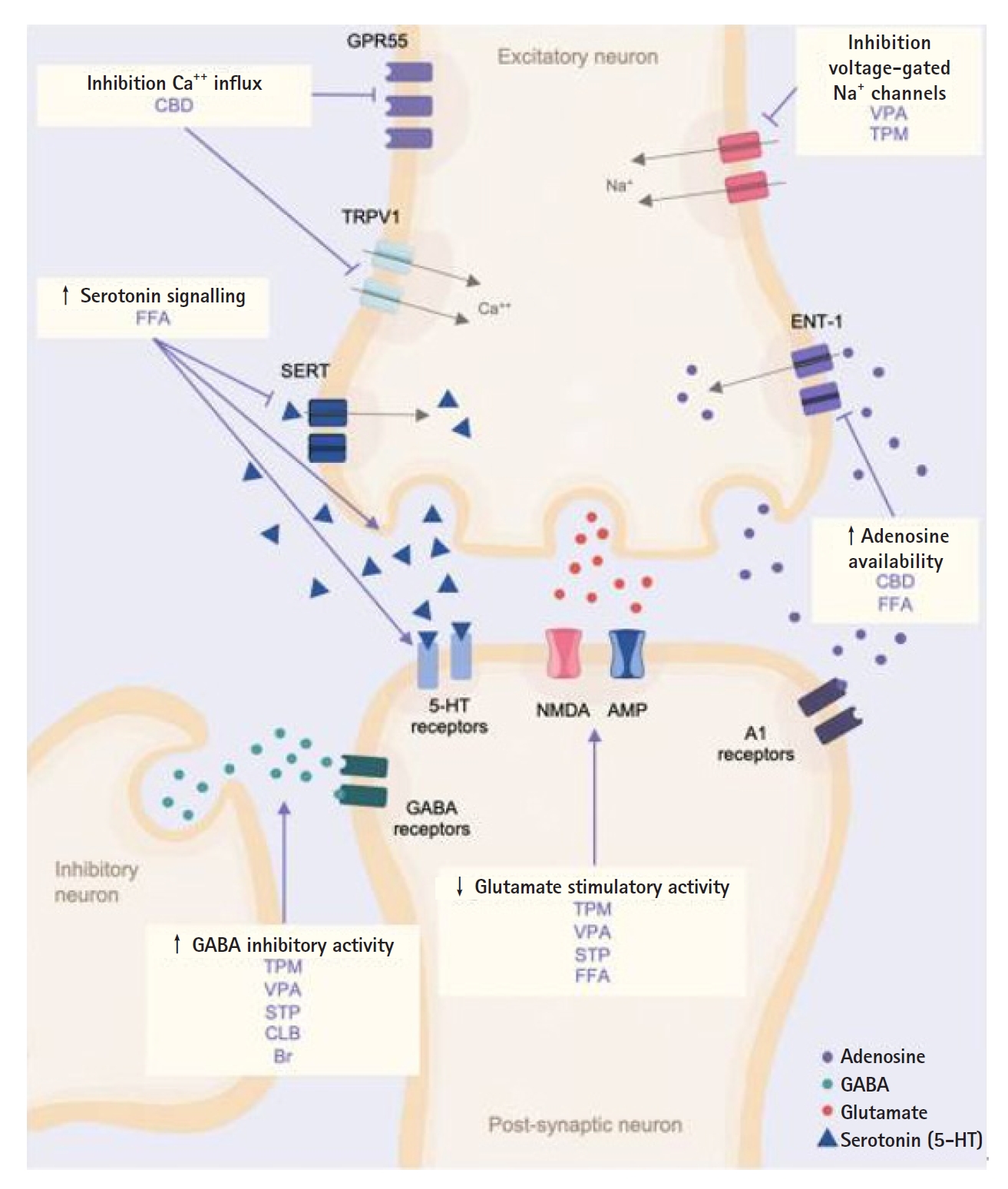
Simplified schematic of the main known mechanisms of action of the antiseizure medications used in Dravet syndrome. GRP55 G, protein-coupled receptor 55; Ca++, calcium; CBD, cannabidiol; Na+, sodium; VPA, valproate, ↑, increased; ↓, decreased [4]; TPM, topiramate; TRPV1, transient receptor potential vanilloid subtype 1; FFA, fenfluramine; SERT, serotonin transporter; ENT-1, equilibrative nucleoside transporter; 1; 5-HT, 5-hydroxytryptamine (serotonin); NMDA, N-methyl-D-aspartate; AMP, alphaamino-3-hydroxy-5-methyl-4-isoxazole propionic acid; GABA, gamma-aminobutyric acid; STP, stiripentol; CLB, clobazam; Br, bromide.
The authors first identified all known genes associated with monogenic epilepsies, including DS, and their corresponding mutations. They then constructed computational models that simulated the effects of each mutation on gene function and predicted how different drugs would interact with the mutated protein. The authors then validated their predictions using experimental data from preclinical models and clinical studies.
Using this approach, the authors identified several drugs that could potentially be effective in treating DS, including sodium channel blockers, AEDs, and drugs that target specific cellular pathways affected by the mutation. Importantly, the authors also identified potential side effects associated with each drug and proposed strategies to mitigate these effects.
In recent years, significant progress has been made in understanding and treating DS. Various therapeutic approaches have been explored to improve the quality of life for individuals with DS. Next, we will discuss several key therapeutic strategies based on the provided references.
1. Stiripentol
Stiripentol has gained approval as an adjunctive therapy for DS in various regions, including Europe, Canada, Japan, and the USA. Its effectiveness was initially explored in a large open-label study involving children and adolescents with refractory epilepsy. Subsequent controlled trials demonstrated its significant efficacy in reducing seizure frequency, with response rates of 71% in one study and 67% in another when added to valproate and clobazam therapy [88]. Meta-analyses further confirmed its superiority over placebo in terms of the response rate and seizure freedom. Long-term observational studies indicated sustained efficacy, with seizure reduction rates in the range of 48% to 63%, along with reductions in the frequency of prolonged seizures, hospitalizations, and the use of rescue medication [89,90]. Importantly, stiripentol's adverse effects, such as drowsiness and loss of appetite, were manageable through dosage adjustments, and discontinuation due to side effects was rare. Stiripentol is often used in combination with other AEDs, notably clobazam, to control seizures in patients with DS [91]. Its mechanism of action involves reinforcing the inhibitory effects of GABAergic neurotransmission. Several studies have suggested that the addition of stiripentol to existing treatments can significantly reduce the frequency of epileptic seizures. This reduction in seizures may improve the quality of life of patients and their families, reducing the burden associated with the condition.
Pharmacokinetic studies showed that stiripentol is rapidly absorbed, metabolized mainly by CYP450 enzymes, and exhibits nonlinear, dose-dependent pharmacokinetics. The relationship between dosage, plasma concentration, efficacy, and safety remains to be fully elucidated, but preliminary findings suggest that factors such as body weight and comedications can impact plasma concentrations. Stiripentol also acts as a potent inhibitor of certain CYP enzymes, notably CYP2C19 and CYP3A4, affecting the metabolism of coadministered AEDs. This can lead to increased plasma concentrations of these drugs, with dosage adjustments necessary to manage adverse effects, particularly somnolence. In cases where somnolence persists, further dose reductions and adjustments of other sedative potential drugs may be considered, with a focus on balancing improved tolerability and seizure control. Stiripentol's complex pharmacokinetic interactions and its role as an effective therapy for DS underscore the importance of careful monitoring and individualized treatment strategies in clinical practice [92].
2. Fenfluramine
Fenfluramine, once used in combination with phentermine as an appetite suppressant, was withdrawn from the market in 1997 due to severe cardiac side effects, including valvular hypertrophy and pulmonary hypertension when administered at high dosages [93]. However, its unique pharmacological properties, such as its high affinity for serotonin receptors (5-hydroxytryptamine [serotonin] 2A and 2C) and modulation of the sigma 1 receptor, led to interest in its potential as an AED [94]. Early case reports and subsequent studies demonstrated the efficacy of low-dose fenfluramine in controlling drug-resistant seizures, particularly in children with intellectual disabilities and early-onset epilepsy, many of whom were later confirmed to have DS. Despite the global ban, Belgium permitted its use in a limited number of DS patients, resulting in long-term follow-up studies showing sustained seizure freedom in the majority of cases [95,96]. Recent prospective, double-blind, placebo-controlled trials further affirmed the safety and efficacy of low-dose fenfluramine in DS patients, both those not taking stiripentol and those on it. These trials demonstrated significant seizure reduction, with minimal side effects and no cardiac complications, leading to the approval of low-dose fenfluramine as a valuable addition to the therapeutic arsenal for DS.
In these trials, fenfluramine showed a substantial improvement in the percentage of responders, with some children becoming seizure-free or experiencing a significant reduction in seizures. Appetite-related issues were observed but generally did not result in significant weight loss. Importantly, rigorous cardiac monitoring throughout the studies did not reveal any cardiac problems associated with fenfluramine use. Long-term extension studies further indicated the stability of its efficacy over more than a year of follow-up, with a substantial reduction in monthly convulsive seizure frequency. This evidence underscores fenfluramine's role as a promising and well-tolerated AED for DS, offering new hope to patients with this challenging epilepsy syndrome [97].
3. Cannabidiol
The therapeutic use of CBD in DS has gained substantial attention. CBD is a non-psychoactive compound derived from cannabis plants. Several studies [25,98], have reported that CBD use led to a significant reduction in seizure frequency in patients with DS. CBD is believed to exert its antiepileptic effects through multiple mechanisms, including modulation of neuronal excitability and inflammation. CBD has been shown to have anticonvulsant properties in preclinical and clinical studies, and it may have neuroprotective effects by reducing oxidative stress and inflammation in the brain. It offers a potential alternative for individuals who do not respond to conventional AEDs.
The most common side effects of CBD treatment are somnolence, decreased appetite, diarrhea, and increased serum aminotransferase concentrations. However, these side effects are generally mild to moderate in severity and tend to decrease over time. Additionally, CBD has a favorable safety profile, and there is no evidence of significant adverse effects on cognitive function or mood [99].
Conclusion and Prospects
The prospects for treating DS, a severe and refractory form of epilepsy, have substantially advanced in recent years. As our understanding of the genetic and molecular basis of the disorder has deepened, targeted therapies are emerging as promising avenues. One of the notable approaches involves sodium channel modulation, since mutations in the SCN1A gene, encoding the Nav1.1 sodium channel, are frequently implicated in DS. Pharmacological agents aiming to restore proper sodium channel function or alternative channel-targeting therapies are under investigation. Additionally, advancements in precision medicine and individualized treatment plans, informed by genetic testing, offer hope for more effective and tailored interventions. Moreover, ongoing research into anti-inflammatory and neuroprotective strategies recognizes the complex interplay of factors contributing to DS. Collaborative efforts between clinicians, geneticists, and pharmaceutical researchers hold the promise of not only ameliorating symptoms but potentially altering the course of the disease itself, providing a brighter outlook for individuals affected by DS and their families.
Notes
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: HEM and MJ. Formal analysis: AN, LB, and SC. Methodology: HEM, AN, and SC. Visualization: SC. Writing-original draft: HEM. Writing-review & editing: HEM, MA, LB, and SC.