Biotinidase deficiency is a rare, autosomal recessive metabolic disorder with a global incidence of 1 in 60,000 births. The clinical manifestations of biotinidase deficiency are varied, impacting the neurological, dermatological, respiratory, and immune systems. Patients with this deficiency may exhibit neurological symptoms such as seizures, hypotonia, sensorineural hearing loss, and optic nerve atrophy [1]. Administering a high dose of biotin can alleviate many symptoms, including convulsions and skin manifestations [2]. However, biotin treatment cannot reverse hearing loss, optic nerve atrophy, or significant developmental delay [3]. Therefore, early detection and treatment are crucial for managing this condition.
In this report, we present a rare case of a 4-month-old girl with biotinidase deficiency. Her initial presentation was an uncontrolled generalized tonic seizure, despite receiving multiple antiseizure medications (ASMs).
A 4-month-old girl was referred to our emergency department after suffering from five instances of generalized tonic seizures. She was the second child of unrelated Korean parents, born at 38 weeks' gestation and weighing 3,500 g at birth. Shortly after her birth, she experienced an episode of apnea and was admitted to the neonatal intensive care unit for a week due to respiratory distress.
Upon admission, the patient demonstrated control over her head movements and the ability to track moving objects. A neurologic examination revealed normal deep tendon reflexes and muscle tone. Further examination of other bodily systems detected audible stridor during breastfeeding. There were no signs of alopecia or other skin manifestations, such as rashes.
The patient's serum lactate level was slightly elevated, at 3.7mmol/L (normal range, 0.36 to 0.75) and ammonia was elevated at 80 μmol/L (normal range, 11 to 30). Mild anemia was present, with a hemoglobin level of 9.6 g/dL (normal range, 10.0 to 14.0), but other parameters in the complete blood count and C-reactive protein levels were normal. Liver function, serum electrolyte levels, and thyroid function were normal. Urine organic acid analysis revealed elevated levels of 3-hydroxyisovaleric acid, at 53.8 mmol/mol creatinine (normal range, 1 to 20) and 3-hydroxyglutaric acid (17.9 mmol/mol creatinine; normal value, 0 mmol/mol creatinine). Serum amino acid analysis revealed no abnormal findings.
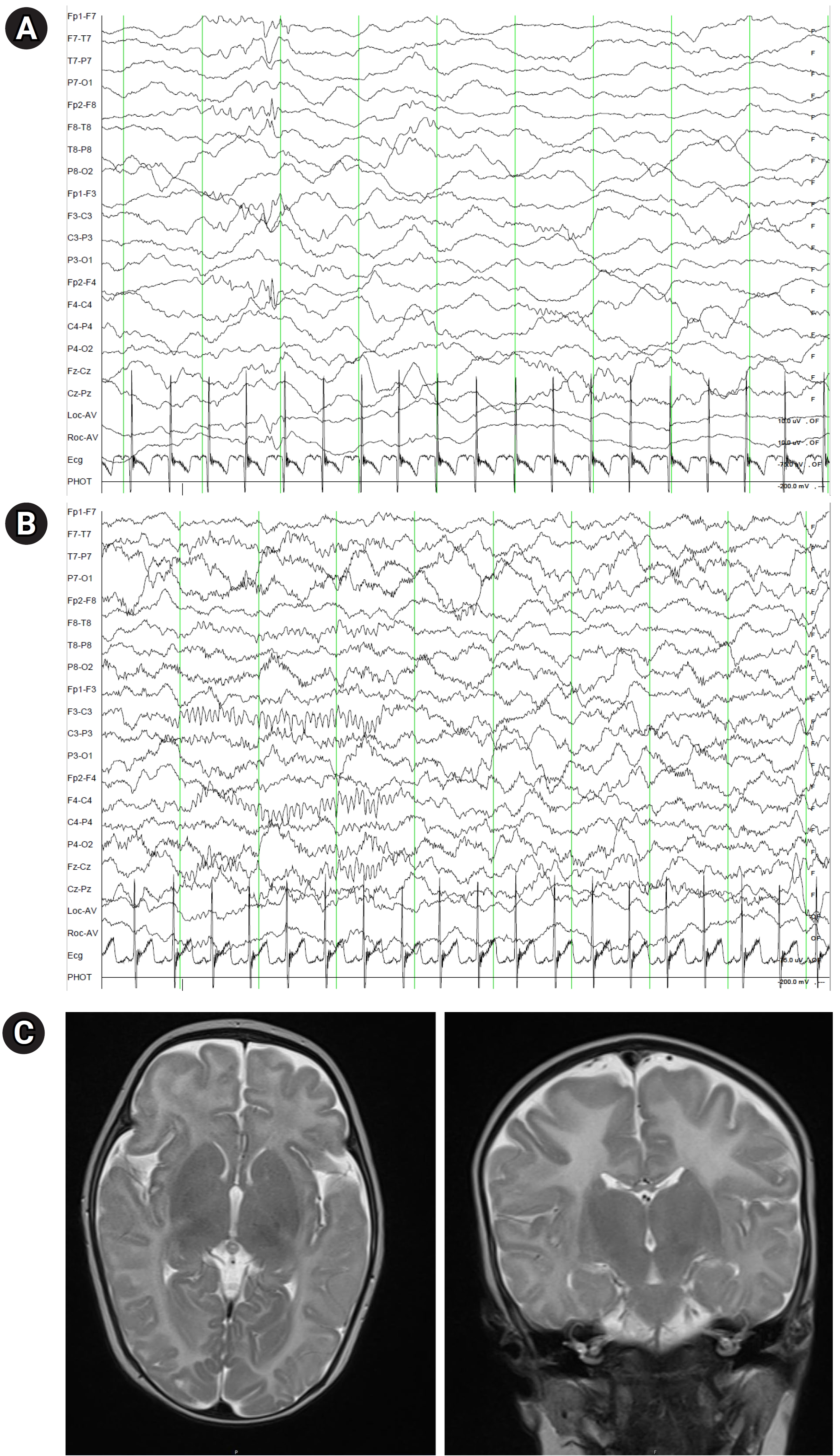
On the 83rd day after birth, the patient underwent an electroencephalogram while sedated and not on ASMs. During sleep, intermittent spike discharges were noted from the bi-frontal areas (Fig. 1A). Brain magnetic resonance imaging showed a pronounced T2 hyperintensity in the supratentorial white matter, especially in the centrum semioval (Fig. 1C).
Her seizures were refractory to multiple ASMs, including levetiracetam, topiramate, valproic acid, and phenobarbital. One month after admission, she exhibited hypotonia and developmental regression. Whole exome sequencing revealed a homozygous variant in the biotinidase (BTD) gene on chromosome 3, characterized by a c.941_942del (p.lle314SerfsTer19) mutation. Subsequent parental genetic testing revealed that her father was heterozygous for a mutation in the same gene, whereas her mother was not. The diagnosis was confirmed through a biotinidase enzyme activity assay test, which revealed no biotinidase activity (0 nmol/min/mL [normal range, 5 to 9]).
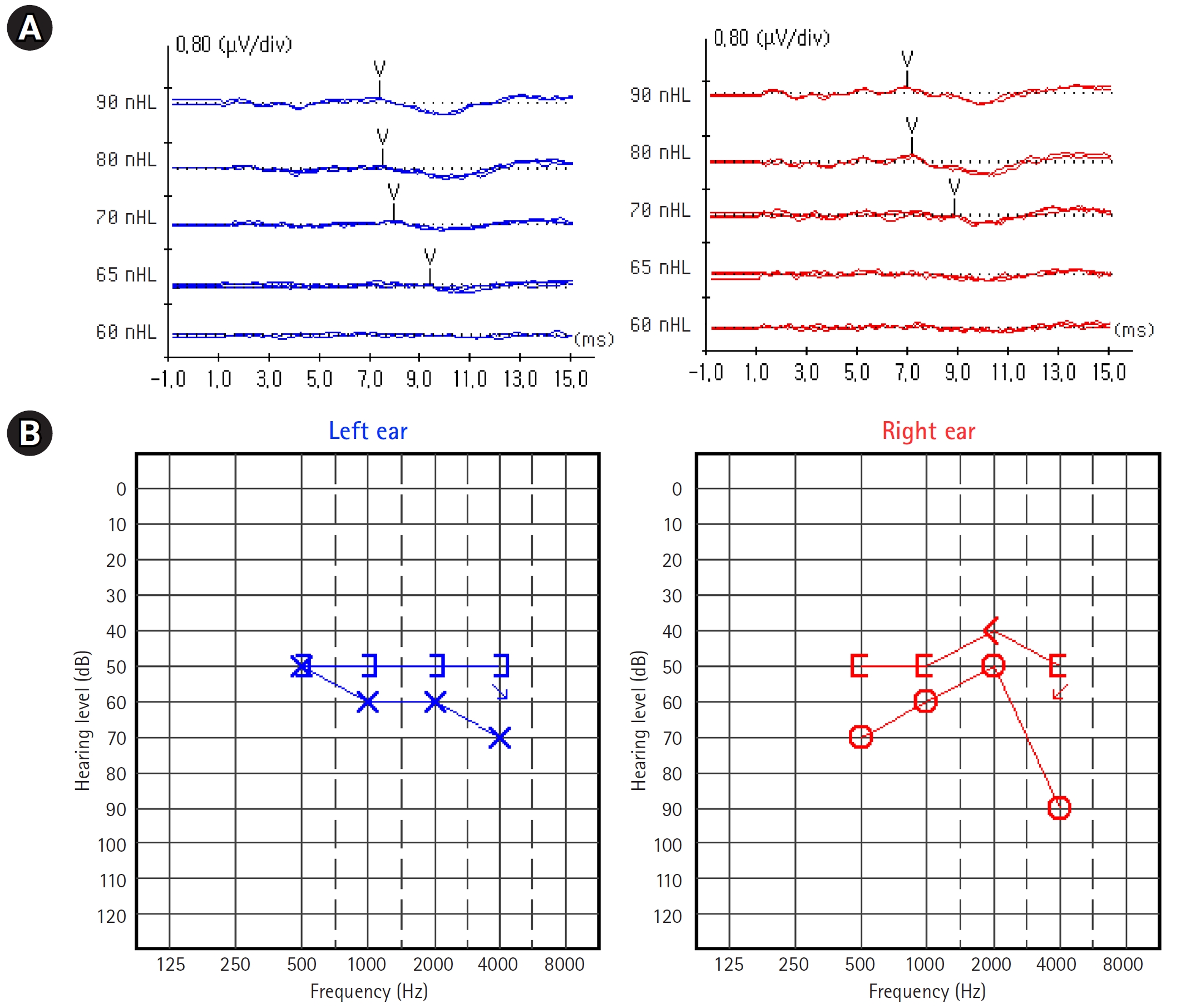
Seizures ceased promptly following the daily administration of 5 mg of biotin, which facilitated the discontinuation of the ASMs. Electroencephalography obtained 4 months after biotin administration revealed a normal sedated sleep record (Fig. 1B). However, despite this treatment, her sensorineural hearing loss and global developmental delay persisted at 9 months of age (Fig. 2). The Denver developmental screening test, conducted when she was 9 months old, indicated that her gross motor skills were equivalent to those of a 6-month-old. Her language, personal-social, and fine motor adaptive skills were at the levels of a 6-, 7-, and 8-month-old, respectively.
Biotinidase is an enzyme that facilitates the cleavage of biotin from biocytin. Biotin, in its free form, serves as a coenzyme for four carboxylases: 3-methylcrotonyl-CoA carboxylase, pyruvate carboxylase, acetyl-CoA carboxylase, and propionyl-CoA carboxylase. These carboxylases are essential for gluconeogenesis, amino acid catabolism, and fatty acid synthesis. Individuals with a deficiency in biotinidase exhibit reduced activity of biotin-dependent carboxylases, which can lead to ketoacidosis and hyperammonemia due to the buildup of abnormal organic acid metabolites. Analysis of urine organic acid can reveal elevated levels of 3-hydroxyisovaleric, 3-hydroxypropionic, and lactic acids, as well as 3-methylcrotonylglyceine in untreated patients [4]. The accumulation of these abnormal and potentially harmful metabolites can result in neurological impairment in patients with biotinidase deficiency [5].
Individuals with profound biotinidase deficiency, which is defined as less than 10% of the average serum biotinidase activity, typically display one or more symptoms if they do not receive treatment [1]. These symptoms can range from neurological issues (including seizures, hypotonia, developmental delays, sensorineural hearing loss, and optic nerve atrophy) to skin conditions (such as eczematous skin dermatitis and alopecia). Respiratory abnormalities like hyperventilation, stridor, and apnea may also be present [6]. Children who have untreated partial biotinidase deficiency may exhibit milder symptoms, especially when exposed to stressors like infection or fasting [7].
Seizures and hypotonia are the most frequently seen neurological symptoms of untreated profound biotinidase deficiency [1]. Roughly 70% of children with profound biotinidase deficiency who show symptoms experience seizures. As demonstrated in our case, an ASMs often fail to control these seizures [5]. However, in the majority of cases, seizures have been noted to stop within hours or days following the administration of biotin [2]. Given that biotin is a water-soluble B-complex vitamin found in various foods and is not linked to toxicity, a trial of biotin should be considered for patients whose seizures are resistant to ASMs.
In 1984, Virginia initiated the inaugural newborn screening program for biotinidase deficiency [8]. This deficiency meets the critical criteria for inclusion in neonatal screening programs. Firstly, the disorder is linked with high morbidity and, if not treated, can result in coma or even death [9]. Furthermore, symptoms such as optic nerve atrophy and hearing loss seem to be less irreversible after biotin administration compared to other symptoms like seizures and dermatitis [5]. Additionally, patients who developed symptoms before diagnosis and treatment may suffer from physical and cognitive developmental delays [3]. Therefore, early newborn screening is vital to identify patients and begin biotin replacement before symptoms appear. Secondly, there are other simple, direct, and effective treatment options available. A daily dose of 5 to 20 mg of biotin replacement is typically recommended. Thirdly, screening methods for biotinidase activity using colorimetric or fluorometric assays are relatively inexpensive compared to tandem mass spectroscopy [8]. Vallejo-Torres et al. [10] conducted an analysis of the cost-effectiveness of a national newborn screening program for biotinidase deficiency in Spain. Considering these factors, the Republic of Korea should contemplate implementing neonatal screening programs for biotinidase deficiency.
This study was approved by the Institutional Review Board of Asan Medical Center (IRB file No. 2023-0687). Written informed consent by the patients was waived due to a retrospective nature of our study.