Introduction
Teenagers with epilepsy are especially prone to physical, cognitive, emotional, and social difficulties, since adolescence is a critical period where autonomy and independence develop, and coping with adulthood is often challenging, even for healthy teenagers [1,2]. Epilepsy and its treatment can have a direct impact on adolescentsŌĆÖ lifestyles, such as education, employment, use of alcohol, contraception, pregnancy, driving ability, and relationships, and can adversely affect their self-esteem and identity. In addition, even minimal side effects of treatment or comorbidities can permanently harm adolescentsŌĆÖ social outcomes; thus, social factors should be considered as particularly important in adolescence [3]. The transition from a pediatric to an adult clinic is also a particular problem at this age [4]. Transition is defined as a purposeful, planned process that prepares adolescents and their families to use the adult-oriented health care system appropriately. However, several barriers, such as the reluctance of families and the complexity of the process, can interrupt this transition, resulting in many patients being lost to follow-up with resultant poor outcomes [5].
Epilepsy occurring during adolescence has been considered to have a relatively benign course and a low incidence of neuropsychiatric comorbidities, as many of these cases have an unknown etiology. In addition, idiopathic generalized epilepsies (IGEs)ŌĆönamely, juvenile myoclonic epilepsy (JME), juvenile absence epilepsy (JAE), and generalized tonic-clonic seizures (GTCS) aloneŌĆöare the specific epilepsy syndromes frequently observed in adolescence, with two-thirds of patients ultimately achieving freedom from seizures and fewer than 20% still having active epilepsy after withdrawal of drugs [6]. These well-characterized syndromes share several commonalities such as generalized seizure types, similar electrographic findings, unremarkable neuroimaging, normal intellectual function, and a presumed genetic etiology [7,8], which has partially helped to understand the profile and natural history of adolescent-onset epilepsy.
Nevertheless, most previous studies focused primarily on specific syndromes, causing limitations in evaluating the prognosis of overall adolescent-onset epilepsy, including other types of epilepsy that are not categorized as epilepsy syndromes [9,10]. Moreover, only a few observational studies have investigated the long-term course and outcomes of epilepsy in adolescence, despite the high incidence of important issues associated with adolescents with epilepsy. This retrospective study aimed to evaluate the long-term pharmacological and psychosocial outcomes of adolescent-onset epilepsy patients followed up for 10 or more years at a tertiary center.
Materials and Methods
1. Patients and data collection
This study identified 1,222 patients diagnosed with epilepsy at the age of 13 to 19 years between April 1993 and April 2019 at the Pediatric Neurology Department of Asan Medical Center. Among them, 137 patients followed up consecutively for at least 10 years were included, and the remaining patients were excluded due to persistent poor adherence to treatment, loss to follow-up, incomplete data, or death (Fig. 1). We retrieved all case histories of patients with adolescent-onset epilepsy from the electronic medical records and evaluated long-term seizure and psychosocial outcomes. PatientsŌĆÖ medical records were retrospectively reviewed regarding sex, birth history, previous history of febrile seizure or status epilepticus, family history of epilepsy, underlying diseases, seizure frequency, epilepsy type, etiology of epilepsy, the presence of neurological deficits or intellectual disability (ID), psychological consequences, socioeconomic status, and transition outcomes. Information on electroencephalography (EEG) and magnetic resonance imaging (MRI) and patientsŌĆÖ history of anti-seizure drug (ASD) use were also collected. Additionally, we searched for possible predictors of epilepsy remission, intractability, and relapse, including sex, history of febrile seizures or status epilepticus, family history of epilepsy, seizure frequency at 3 months and 1 year after initial treatment, etiology of epilepsy, any abnormalities on EEG or brain MRI, the presence of neuropsychiatric comorbidities, and compliance, as previously described. This study was approved by the Medical Sciences Ethics Committee of Asan Medical Center (IRB file No. 2021-0285). Due to a retrospective nature of our study, the requirement for written informed consent from the patients was waived.
2. Definitions
The terminology and classification of seizures and epilepsies were defined according to the latest International League Against Epilepsy (ILAE) recommendations [11]. Epilepsy was classified as focal, generalized, combined focal and generalized, and unknown. An epilepsy syndrome was diagnosed when a patient represented a cluster of age-dependent features occurring together with distinctive seizure types, EEG findings, and developmental status. The term ŌĆ£IGEsŌĆØ was used to encompass four epilepsy syndromes: childhood absence epilepsy, JAE, JME, and GTCS alone [7]. The etiology was classified as structural, genetic, metabolic, infectious, autoimmune, and unknown. Each patientŌĆÖs epilepsy was classified into one etiologic category, which was considered as the most likely putative cause of epilepsy.
Seizure outcomes were evaluated based on the seizure-free period at the last follow-up [12]. Remission of epilepsy was defined as a patient achieving 2-year seizure freedom during the follow-up. Two-year or longer periods of remission at the end of follow-up, regardless of the use of ASDs, were referred to as terminal remission (TR). Resolution of epilepsy was defined as a seizure-free period of at least 5 years or longer after the withdrawal of ASD during a sustained remission. Relapse represented any subsequent seizures following remission. Intractability was designated as a failure to achieve remission despite adequate trials of two appropriately chosen ASDs.
Long-term psychological and socioeconomic consequences were analyzed with respect to psychiatric disorders, educational level, and employment status based on reports from the patients and their caregivers. Educational status was defined as the highest level accomplished by the patients until the end of follow-up and categorized into four levels: college, high school, elementary school, and special school (skilled). Employment status was categorized into three classes: full-time job, part-time job, and unemployed.
Transition is a complex and multidirectional process that prepares adolescents and their families to use adult health clinics appropriately. Adolescents with epilepsy who transitioned from pediatric to adult care were investigated for their clinical features, seizures, neuropsychiatric outcomes, and possible associated factors.
3. Statistical analysis
Data processing and analysis were performed using SPSS version 18.0 (SPSS Inc., Chicago, IL, USA). We used the chi-square test to analyze possible differences between patients with or without completely resolved symptoms at the end of follow-up. Bivariate logistic regression analysis was also conducted to calculate odds ratios (ORs) with 95% confidence intervals (CIs) and identify predictive factors for seizure outcomes. Statistical significance was defined as a P<0.05.
Results
A total of 137 (86 male) patients who were diagnosed with epilepsy in adolescence and followed up for more than 10 years were included in this study. The median age at epilepsy onset was 14 years, and the mean follow-up duration was 14.2 years (range, 10 to 25). All the patients were born full-term and only one (0.7%) patient experienced hypoxic-ischemic encephalopathy. Nineteen (13.9%) patients had a previous history of febrile seizures and 13 (9.5%) patients had a family history of epilepsy. Status epilepticus was observed in five (3.6%) patients. Ten (7.3%) patients had underlying diseases: three with primary brain tumors, two with hematologic malignancies including acute lymphoblastic leukemia and non-Hodgkin lymphoma, two with end-stage renal disease, one with congenital heart disease, one with acute liver failure, and one with 22q11.2 deletion syndrome. According to the new proposal for epilepsy classification (ILAE, 2017), epilepsy was classified as focal in 62 (45.3%) patients, generalized in 55 (40.1%), unknown in 11 (8.0%), and combined generalized and focal in nine (6.6%). Fifty (36.7%) patients had epilepsy syndromes, most of whom (48/50, 96.0%) were diagnosed with IGEs: 24 (17.5% of total) patients had epilepsy with GTCS alone (EGMA); 20 (14.6%) patients had JME; and four (2.9%) patients had JAE. The remaining two (1.5%) patients had developmental and epileptic encephalopathy. The epilepsy etiology was classified as unknown (57/137, 41.6%), genetic (50/137, 36.5%), structural (24/137, 17.5%), and infectious causes (6/139, 4.4%). Brain MRI was performed in 131 (95.6%) patients and pathological findings were found in 25 (18.2%) patients. The most common structural abnormality was hypoxic-ischemic encephalopathy, in 8/25 (32.0%) of patients, followed by focal cortical dysplasia in 6/25 (24.0%), hippocampal sclerosis in 4/25 (16.0%), brain tumors (three with ganglioglioma and one with central nervous system involvement of leukemia) in 4/25 (16.0%), and intracranial hemorrhage in 3/25 (12.0%). No patient with adolescent-onset epilepsy died during the follow-up. Details of the demographic and clinical features, underlying diseases, and etiologies are presented in Table 1.
1. Seizure outcomes and treatment
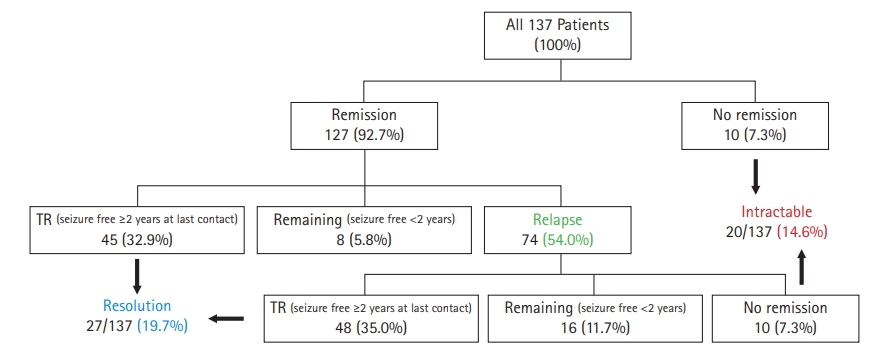
During follow-up, 127 (92.7%) patients experienced at least one remission period, of whom 93 (67.9% of total) patients achieved TR at last contact. Epilepsy resolved in 27 (19.7%) patients. Of patients who had a first remission, 74 (54.0% of total) patients experienced a relapse. Forty-eight (35.0% of total) patients finally achieved TR, while 10 (7.3%) patients still had active epilepsy, and 16 (11.7%) remained seizure-free for less than 2 consecutive years. Of the 44 patients who never reached TR at the end of study, 20 (14.6%) patients were found to have intractable epilepsy. In a subgroup analysis of epilepsy syndrome, most of the IGE patients (46/48, 95.8%) achieved remission at least once during the follow-up period, but 28 (58.3%) of them relapsed, and only six (12.5%) finally reached resolution. Intractable epilepsy was observed in six (12.5%) patients among those diagnosed with IGEs. Fig. 2 demonstrates the patientsŌĆÖ characteristics and outcome chart.
Among the 137 final ASD therapies, 48 (35.0%) regimens were monotherapy and 89 (65.0%) were polytherapy (Ōēź2 ASDs). Approximately 60% (29/48) of the IGE patients needed polytherapy to achieve freedom from seizures. The median number of ASDs used was 2 (range, 1 to 10), and the mean duration of therapy was 13.0 years (median, 12.3; range, 1.0 to 25.9). Of the 93 patients included in the TR group, 42 (30.7%) achieved freedom from seizures by taking monotherapy, while the remaining 51 (37.2%) were controlled on a combination of two to five ASDs. The patients who never achieved TR (44/137, 32.1%) used more drugs, with a mean number of 3.9 ASDs which was significantly higher than that of the TR group (mean, 1.8; P=0.000). Valproic acid was the most frequent drug administered, in 80 (58.4%) patients, followed by lamotrigine in 54 (39.4%) patients, topiramate in 43 (31.4%) patients, and oxcarbazepine in 36 (26.3%) patients. Poor adherence was observed in 65 (47.4%) patients. In addition to ASD treatment, six (4.4%) patients received palliative therapies for uncontrolled seizures. A ketogenic diet and palliative surgery were performed in five (3.6% of total) patients and in one (0.7%) patient, respectively.
2. Factors associated with seizure outcomes
A univariate analysis showed no statistically significant differences between those who achieved TR and those who did not in terms of sex, history of perinatal distress, febrile seizure, status epilepticus, family history of epilepsy, EEG abnormalities at least once during the follow-up, structural abnormalities, epilepsy types, etiologies of epilepsy, psychiatric comorbidities, and medication adherence (Table 2). However, seizure frequency at 3 months (P=0.029) and at 1 year after initial treatment (P=0.001) and absence of ID at seizure onset (P=0.015) were highly associated with TR of epilepsy. Further bivariate logistic regression analysis demonstrated that seizure freedom at 1 year (OR, 4.02; 95% CI, 1.71 to 9.41; P=0.001) and normal intellectual function (OR, 3.37; 95% CI, 1.13 to 10.10; P=0.030) tended to increase the chance of achieving TR. In addition, seizure frequency at 1 year after initial treatment (P=0.002) was also a significant predictor of intractability, and patients who continued to experience seizures at 1 year had a higher probability of having intractable epilepsy at the end of the study (OR, 7.98; 95% CI, 1.77 to 35.97; P=0.007). The potential risk factors for relapse included EEG abnormalities at least once during follow-up (OR, 8.23; 95% CI, 1.39 to 48.87; P=0.020), poor compliance (OR, 4.84; 95% CI, 2.13 to 11.02; P=0.000), and history of febrile seizures (OR, 4.10; 95% CI, 1.21 to 13.93; P=0.024) in logistic regression analysis (Table 3).
3. Neurodevelopmental and psychosocial outcomes
Neurodevelopmental comorbidities were documented in 17 (12.4%) patients. Sixteen (11.7%) patients showed ID, eight of whom had an intelligence quotient below 50. Cerebral palsy (CP) was identified in 5/137 (3.6%) patients. Previous or current psychiatric problems were reported in 12 (8.8%) patients, as follows: behavioral disorders (7/12, 58.3%), depression (5/12, 41.7%), panic or adaptation disorders (3/12, 25.0%), and schizophrenia (2/12, 16.7%). Of the 107 (78.1%) patients who had received formal education, 92 (67.2% of total) had reached college as the highest educational attainment. The remaining patients had graduated with a high school diploma (nine patients, 6.6%) or from a special school (six patients, 4.4%). Employment information was reported in 101 of the 137 patients. At the end of follow-up, 77.2% (78/101) of the patients were employed full-time with sufficient income; 12.9% (13/101) were employed part-time or in manual labor; and 8.9% (9/101) were unemployed or on welfare. Normal intellectual function (OR, 7.83; 95% CI, 2.38 to 25.79; P=0.001) was a significant factor that predicted a higher likelihood of employment in the logistic regression analysis model.
4. Transition from pediatric to adult care
During the follow-up period, 36 (26.3%) patients transitioned from our pediatric neurology clinic to other adult epilepsy centers at a mean age of 21.9 years. Most of them had genetic (15/36, 41.7%) or unknown (13/36, 36.1%) etiologies and showed EEG abnormalities at least once during their follow-up (33/36, 91.7%). An epilepsy syndrome was identified in 15/36 (41.7%): seven with JME, seven with EGMA, and one with JAE. However, only 29.4% (5/17) of the patients with comorbid neurodevelopmental disorders (four with ID and two with CP) and 33.3% (4/12) of those with psychosocial problems (one with a behavioral disorder, one with depression, one with schizophrenia, and one with adaptation disorder) were transitioned to adult clinics by the end of the study. The mean duration of ASD therapy was 14.6 years (median, 13.6; range, 6.6 to 25.9) and polytherapy was administered in 25/36 (69.4%). One patient (2.8%) was administered a ketogenic diet. At the end of the study, TR was achieved in 16/36 (44.4%) patients, only four (11.1%) of whom had complete symptom resolution. Nearly half of the transitioned patients (15/36, 41.7%) were found to have intractable epilepsy at the time of transition. No independent factors were significantly associated with transition on logistic regression analysis.
Discussion
This is a study evaluating the pharmacological and psychosocial outcomes of 137 adolescent-onset epilepsy patients followed up for more than 10 years at a single tertiary center. Most of the patients included in this study had unknown (57/137, 41.6%) or genetic (50/137, 36.5%) etiologies, and only 24 (17.5% of total) showed structural abnormalities on their brain MRI. Focal epilepsy (62/137, 45.3%) was slightly more frequent than generalized epilepsy (55/137, 40.1%). Fifty (36.7%) patients had epilepsy syndromes, of whom 48 (96.0%) patients were diagnosed with IGE, and the remaining two (1.5%) had developmental and epileptic encephalopathy.
In this analysis, a 2-year seizure-free state at last contact was defined as TR. Good overall seizure control was found, with a TR rate of 67.9% (93/137), which was comparable to what was reported (68%) in the 30-year cohort study on teenagers with epilepsy [13]. Two previous studies performed among adolescents and adults diagnosed with epilepsy also demonstrated similar results, with 1-year seizure-free rates 63.7% and 68%, respectively, although there was a difference in the period of remission [14,15]. Moreover, only 14.6% (20/137) of patients had intractable epilepsy at the end of this study. This rate is comparable to the pooled incidence proportion (15%) reported in a recent meta-analysis on drug-resistant epilepsy of children [16,17].
According to the recent meta-analysis of 15 studies performed in children, adolescents, and adults with epilepsy between 2005 and 2019, the relapse rate varied substantially depending on the duration of remission: 12.4% to 35% in cases with a remission period within 12 months, 22.8% to 100% with a remission duration of 2 years or more, and 57% to 64.6% with a remission period of 4 years or more [18]. Other factors significantly associated with seizure relapse were age at onset, etiologies, type of seizures, and EEG abnormalities. Many studies have reported that the highest relapse rate in patients with epilepsy onset occurred during adolescence [19-21]. A high rate of relapse was also observed in this analysis, and almost half of the patients with adolescent-onset epilepsy (74/137, 54.0%) experienced relapse after achieving remission once during follow-up.
The main predictor of seizure relapse in this study was the presence of EEG abnormalities (OR, 8.23; 95% CI, 1.39 to 48.87; P=0.020). It is a well-known fact that the relapse rate is higher in seizure-free patients with interictal epileptiform discharges (IEDs) despite some debates on the role of EEG in predicting the risk of seizure recurrence after ASD withdrawal [22-24]. Rathore et al. [25] observed that a three-fold higher relapse risk occurs after withdrawal in patients with IEDs than those with a normal EEG among the 262 patients with temporal lobe epilepsy who underwent epilepsy surgery, and a large meta-analysis of 2,349 epilepsy patients showed that an abnormal EEG, including paroxysmal slowing or spike and slow-wave discharges, is an important red flag sign for seizure relapse [26]. Additionally, the International Federation of Clinical Neurophysiology guidelines in 2018 [27] demonstrated that the presence of IEDs in patients with controlled seizures predicts a higher risk of relapse after ASD withdrawal. Moreover, the high proportion of patients with IGE included in our cohort could have contributed to this result, since IGE is usually considered a lifelong disorder.
Poor adherence to ASD was another significant risk factor for relapse (OR, 4.84; 95% CI, 2.13 to 11.02; P=0.000) in this report. Of the 65 patients (47.4% of total) with adherence problems, 45 (69.2%) experienced seizure relapse during the follow-up period. Adolescence is a particularly vulnerable period, since adolescents have increased autonomy and independence but poor execution of necessary tasks due to lack of parental supervision, decreased motivation, and increased peer pressure [3,28,29]. This often results in reduced adherence to medical treatment compared to children and adults [1,30], which can cause a variety of negative consequences, including an increased risk of mortality, uncontrolled seizures, and high health care costs. Therefore, patient-oriented education programs, personal circumstances and social support, and technology-focused adherence interventions to promote an understanding of epilepsy and adherence to ASD are considered essential for adolescents with epilepsy.
In the present study, seizures in nearly two-thirds of all patients (89/137, 65.0%) were controlled via polytherapy, and almost half of the patients in the TR group (51/93) needed two or more ASDs for their seizure freedom until the last follow-up. In addition, only a small percentage of the TR group (27/93) finally reached resolution at the end of this study, implying a relatively low rate of sustained remission after ASD withdrawal compared to that when using ASD. These findings demonstrated that the majority of patients with adolescent-onset epilepsy were responsive to ASD therapy, but showed a high risk of seizure relapse after withdrawal, ultimately necessitating long-term treatment [7,31].
Neuropsychiatric comorbidities are especially common in children with epilepsy and are often known to have a severe impact on their quality of life [32]. In this study, a relatively lower rate of neurodevelopmental (17/137, 12.4%) and psychiatric (12/137, 8.8%) comorbidities was observed compared to that of previous reports on childhood-onset epilepsy [33-35]. This analysis, however, identified that an absence of ID at seizure onset (P=0.015) was highly associated with the remission of epilepsy at last contact, and normal intellectual function (OR, 3.37; 95% CI, 1.13 to 10.10; P=0.030) was a strong predictor of TR. Given this significant association between epilepsy and comorbidities [32,36,37], the management of neurological and psychological comorbidities is also necessary when planning epilepsy treatment.
Evaluation of long-term socioeconomic consequences and management of transition are important issues in both childhood- and adolescent-onset epilepsy. Nevertheless, only 70% to 80% of children with epilepsy received compulsory education due to negative attitudes or stigma in the past [38]. In addition, the employability rate has generally been reported as only approximately 70% among epilepsy patients in more recent studies [39], and Jennum et al. [40] showed that the rate was much lower among people with epilepsy than in controls in their prospective cohort study. The patients included in this study, however, showed higher educational levels and employability rates. These findings are probably explained by the overall good seizure control and low incidence of neurological comorbidities among patients with adolescent-onset epilepsy.
At the end of this study, 36 (26.3%) patients were transitioned to an adult epilepsy center, only four (11.1%) of them achieved resolution, and nearly half of them (15/36, 41/7%) were found to have intractable epilepsy. It is thought that the transition in our center was mainly focused on patients with poor seizure control necessitating long-term ASD treatment. Most of the patients and their family members, conversely, refused to leave pediatricians with whom they had become familiar. Although transition care for adolescents with epilepsy is considered beneficial, several patient- and clinician-related barriers still constrain the process of transition, as observed at our center. To prevent irreversible medical and social problems caused by discontinuity of chronic care, early interventions with medical and non-medical support tailored to patientsŌĆÖ specific health needs and an organized transitional service will be required for optimal transition care.
A strength of our work is that this is a long-term follow-up study evaluating both the pharmacological and psychosocial outcomes of adolescent-onset epilepsy. In addition, all patients investigated in this study comprised a uniform group of epilepsy patients treated at a single epilepsy center, which could increase the internal validity of the data. However, concurrently, this single tertiary center-based study has limitations in terms of the generalizability of the results, including selection bias for more severely affected cases.
In this study, adolescent-onset epilepsy showed a relatively high TR rate and low rate of intractability, indicating overall favorable seizure outcomes. Frequent relapse after the withdrawal of ASDs, however, required long-term treatment using multiple drugs, and in particular, the presence of any EEG abnormalities and poor adherence to ASD were identified as significant factors in predicting relapse. Additionally, adolescence is a transitional period and adolescents are especially vulnerable to cognitive, emotional, physical, and social difficulties; hence, chronic conditions such as epilepsy can cause a variety of negative impacts on personal and social domains in adolescents. Therefore, a comprehensive treatment plan encompassing not only complete seizure control, but also early recognition and intervention for neuropsychiatric comorbidities, socioeconomic status, and transition-related problems is strongly needed for adolescents with epilepsy to improve their long-term quality of life.