Hypochondroplasia with Acanthosis Nigricans and Moyamoya Disease
Article information

Mutations in the gene encoding fibroblast growth factor receptor 3 (FGFR3) cause a variety of disorders, including achondroplasia, hypochondroplasia (HCH), Crouzon syndrome with acanthosis nigricans (CAN), Muenke syndrome, and thanatophoric dysplasia [1]. Acanthosis nigricans (AN) is a skin condition characterized by velvety and pigmented hyperkeratosis in body folds and creases. It typically affects the neck folds, armpits, groin, and other areas, and is commonly observed in children with obesity, type 2 diabetes, drug-induced conditions, and inherited conditions, including FGFR3 mutations [2,3]. Recent genome-wide association studies have identified the ring finder protein 213 gene (RNF213) as an important susceptibility gene for moyamoya disease (MMD). Here, we describe the first known case of co-occurrence of HCH with AN and MMD, caused by mutations in FGFR3 and RNF213.
An 18-year-old girl was referred to the pediatric department for an evaluation of progressive hyperpigmentation of her skin since the age of 8 years. She was born through normal vaginal delivery at term, and the pregnancy of her mother was uncomplicated. The patient’s family history was unremarkable, motor and intellectual development were normal, and she had not experienced seizures, hemiparesis, or loss of consciousness. On physical examination, she did not have a dysmorphic face, macrocephaly, brachydactyly, lordosis, or a limited range of motion at the elbows. Her skin showed extensive AN at the flexion sites, neck, upper trunk, and groin. Her height was 145.5 cm (0th percentile), and her weight was within the normal range. A skin biopsy revealed no specific findings other than hyperkeratosis. This case was reviewed and approved by the Institutional Review Board of Pusan National University Hospital (IRB No. 2203-007-112). Informed consent was obtained from the parents. The patient’s medical records and other data were anonymized to ensure confidentiality.
Upon an initial analysis of the results of diagnostic exome sequencing (DES), we identified a heterozygous c.1949A>C (p.Lys650Thr) mutation in FGFR3, which has been reported to be associated with CAN and HCH. For further evaluation, a three-dimensional computed tomography (CT) scan of the skull and magnetic resonance imaging (MRI) of the brain were performed. CT showed no craniosynostosis (Fig. 1A). Brain MRI revealed MMD-specific findings, including bilateral distal internal carotid artery stenosis, no visible middle and anterior cerebral arteries, and collateral vessels at the bilateral posterior cerebral arteries and deep gray matter (Fig. 1B and C). We reanalyzed the DES data and identified an additional pathogenic variant of RNF213 (c.14429G>A, p.Arg4810Lys) associated with MMD. Sanger sequencing tests were performed to validate the two variants of FGFR3 and RNF213 in DNA samples from the proband and parents. The parents did not carry FGFR3 c.1949A>C, which had occurred de novo. The patient inherited RNF213 c.14429G>A from her mother (Fig. 1D and E), who was a carrier without MMD on brain MRI.
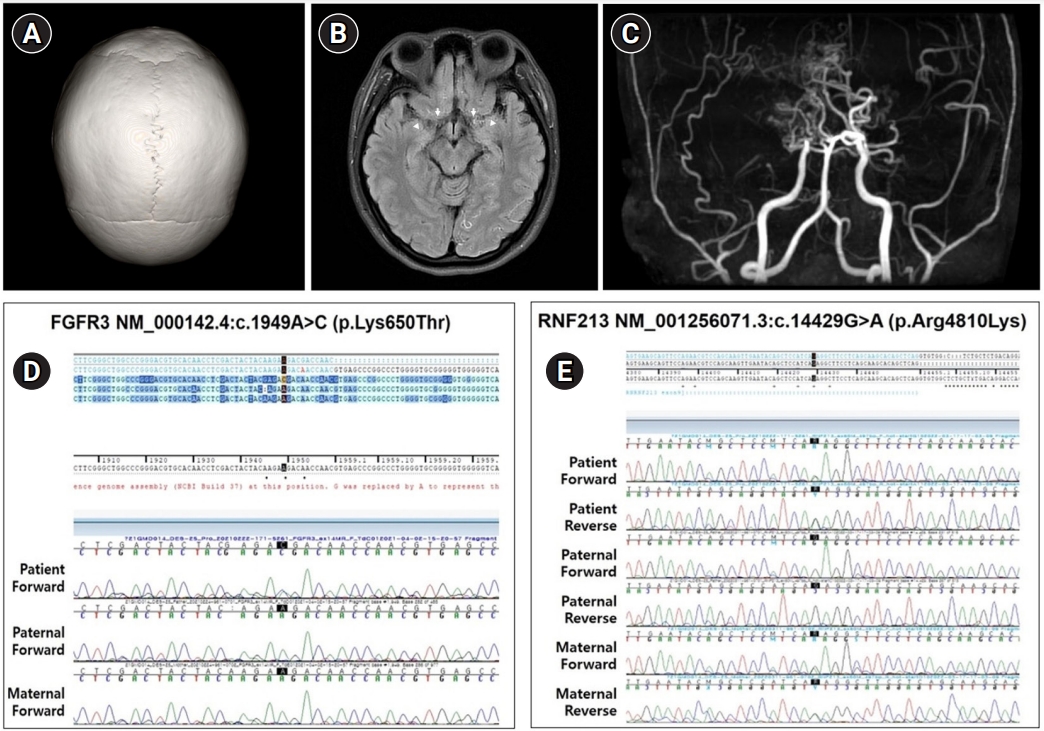
(A) Three-dimensional computed tomography of the skull is normal. (B) T2-weighted axial fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging demonstrates poor visualization of both internal carotid arteries (arrows) and middle cerebral arteries (arrowheads). (C) Magnetic resonance angiography shows bilateral stenosis of the internal carotid, anterior cerebral, and middle cerebral arteries. (D) Sequencing of fibroblast growth factor receptor 3 (FGFR3) identified a heterozygous c.1949A>C mutation in the proband, but not in the parental DNA. (E) Ring finder protein 213 gene (RNF213) sequencing demonstrated a heterozygous c.14429A>C mutation in the proband and the mother’s DNA, but not in the father’s DNA.
HCH is a form of short-limbed dwarfism, like achondroplasia, but with milder features. It is characterized by short stature, short arms and legs, broad, short hands and feet, macrocephaly, lordosis, and a limited range of motion. These symptoms are less prominent than those of achondroplasia and may not be diagnosed until adolescence or adulthood. FGFR3 encodes a transmembrane tyrosine kinase receptor located on the cell surface of fibroblasts and keratinocytes, and is involved in a variety of activities, including mitogenesis, angiogenesis, and wound healing [1,4]. Gain-of-function mutations in FGFR3 result in the activation, proliferation, and differentiation of fibroblasts and keratinocytes, which cause the development of the AN phenotype. To date, several FGFR3 mutations have been reported in patients with HCH. p.Asn540Lys was the most common, and p.Lys650Asn and p.Lys650Gln were associated with a milder skeletal dysplasia phenotype [4]. Only 10 cases have been reported of AN with or without HCH due to the FGFR3 p.Lys650Thr mutation [5-8]. The present patient had the FGFR3 p.Lys650Thr mutation and exhibited AN and a mild HCH phenotype with only short stature and no skeletal dysplasia.
After receiving the first result of the FGFR3 mutation in DES, MMD was diagnosed during the evaluation to confirm deep phenotyping. A reanalysis of the DES data identified a heterozygous c.14429A>C, p.Arg4810Lys mutation in RNF213. MMD is characterized by occlusion of the supraclinoid internal carotid artery and the development of a fine vascular network. Recent advances have enhanced our understanding of the genetic factors associated with MMD. RNF213 has been identified as a novel susceptibility gene for MMD in East Asian people [9]. A case-control study demonstrated a strong association of p.Arg4810Lys with MMD in an East Asian population, with an odds ratio of 111.8 [9]. The frequency of p.Arg4810Lys in the general population in Japan is 1.4% to 2.7%, with incomplete penetrance [10]. A carrier may not exhibit MMD.
To our knowledge, this is the first case of HCH co-occurring with AN and MMD. Additional diagnostic tests or a reanalysis of results is essential for a final and accurate diagnosis, even after a thorough physical examination, phenotyping, and molecular genetic testing. Therefore, to understand the concept of an “evolving” molecular interpretation, education for physicians and patients is essential.
Notes
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: YJL, YHJ, MBK, and YMK. Methodology: YHJ. Project administration: MBK. Writing-original draft: YJL. Writing-review & editing: YMK.
Acknowledgements
This work was supported by a clinical research grant from Pusan National University Hospital in 2022.