Myelin oligodendrocyte glycoprotein (MOG) is an essential component of oligodendrocyte surface membranes [1]. Numerous studies have implicated MOG in immune-mediated demyelinating diseases, including acute disseminated encephalomyelitis, optic neuritis, and transverse myelitis [2]. However, MOG antibody-associated diseases are not limited to demyelinating syndromes. Cerebral cortical encephalitis (CCE), first described by Ogawa et al. [2] in 2017, is a newly identified phenotype of MOG antibody-associated disease. Common symptoms of anti-MOG-associated CCE include seizures, headache, encephalitic features, and cortical symptoms, such as paresis [2]. Unilateral cortical hyperintensities on T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) sequences are a characteristic finding of this phenotype [2]. Viral encephalitis [3], small-vessel vasculitis [4], and hemiplegic migraine [5] should be ruled out, but doing so poses challenges. Furthermore, numerous magnetic resonance imaging (MRI) findings, apart from isolated cortical hyperintensities, have also been reported in anti-MOG-associated encephalitis [6]. Thus, clinicians need to maintain a high index of suspicion for anti-MOG-associated disease when faced with an unusual presentation of encephalitic pathologies, such as unilateral headache with or without transient hemiparesis. Here, we report the diagnostic journey of a girl who was ultimately diagnosed with anti-MOG-associated-CCE. She had two discrete encephalitic episodes 1.5 years apart: the first episode was initially diagnosed as Epstein-Barr virus (EBV) encephalitis and the second was presumed to be a hemiplegic migraine.
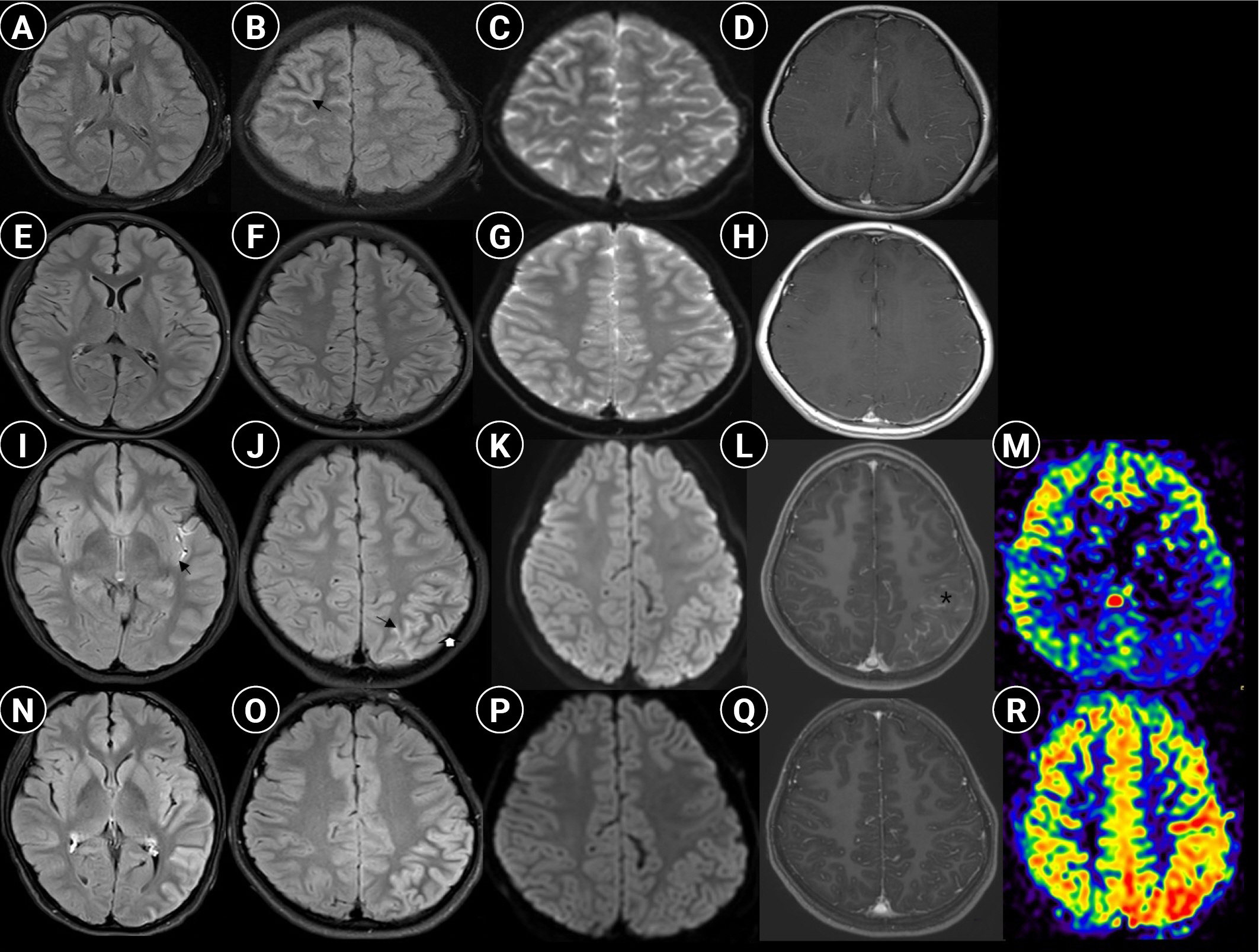
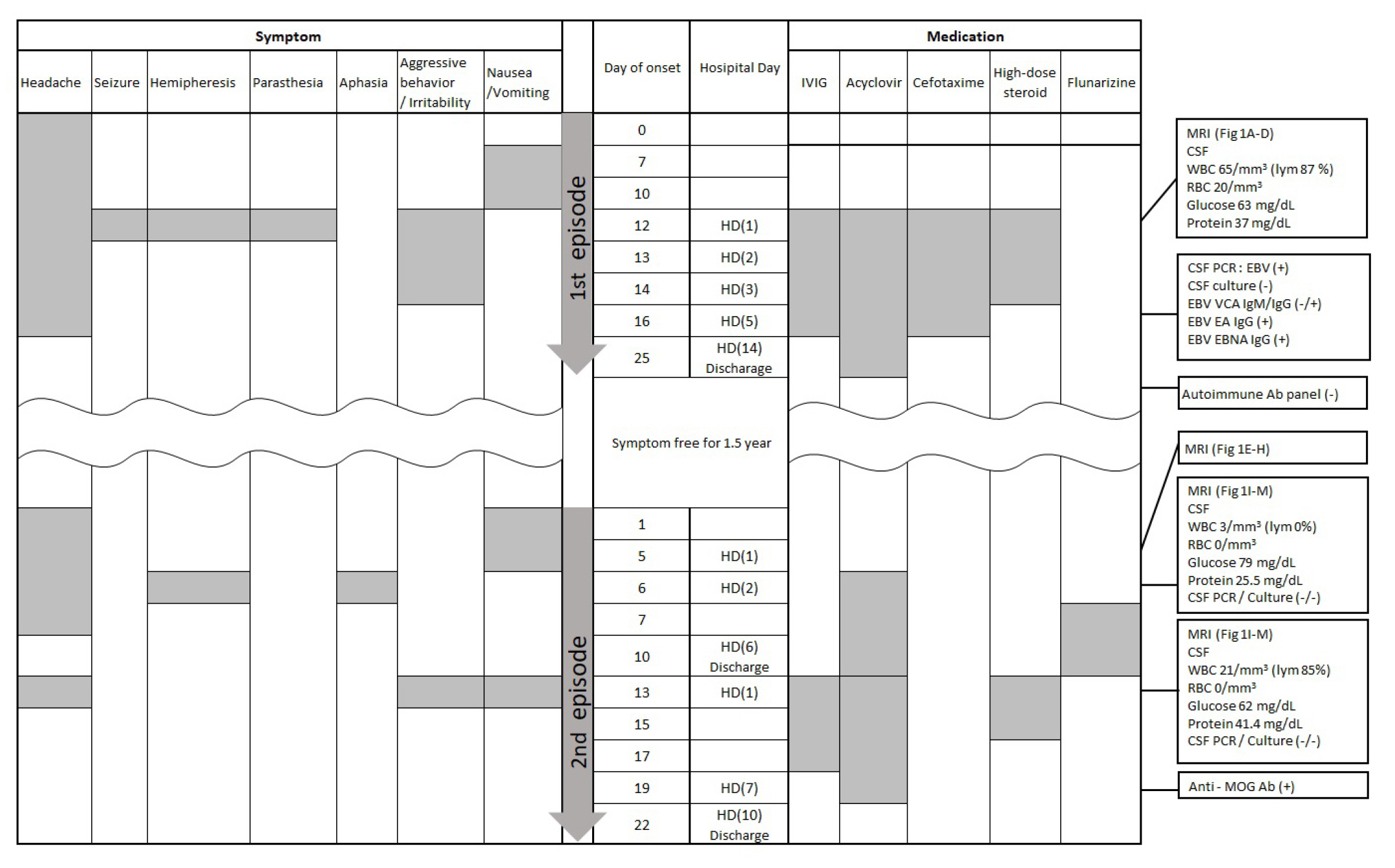
The patient was first admitted at the age of 9 years with headache and repeated seizures. Focal seizures arising from the left side evolved into bilateral tonic-clonic seizures. Ipsilateral weakness and paresthesia persisted for several hours following the seizures. Irritability, aggressive behavior, and severe headaches on the right side of the head were noted. Three days prior to the onset of seizures, she had visited the outpatient clinic due to a headache that lasted 2 weeks. This moderate to severe headache lasted all day, accompanied by vomiting, but improved after sleeping. No focal neurological deficits were noted. Brain MRI and magnetic resonance angiography (MRA) were performed after admission. No abnormality was found on MRA, while sulcal hyperintensity in the right frontoparietal lobes was revealed on MRI (Fig. 1A-D). Subtle cortical hyperintensity was observed on T2-FLAIR sequences, but not on diffusion-weighted imaging. Cerebrospinal fluid (CSF) analysis revealed lymphocyte-predominant mild pleocytosis: white blood cell (WBC) count, 65/mm3; lymphocyte percentage, 87%; red blood cell count, 20/mm3; glucose, 63 mg/dL; and protein, 37 mg/dL. The blood WBC count was slightly elevated, at 13,370/mm3, with neutrophil predominance (68%). All other laboratory findings were normal. Intravenous cefotaxime and acyclovir were administered until central nervous system (CNS) infection was ruled out. Intravenous immunoglobulin and high-dose steroids (30 mg/kg/day for 3 days) were also administered due to the possibility of autoimmune encephalitis since the disease course progressed over 2 weeks without fever. After admission, there were no seizures, and the headache gradually improved. On day 4, EBV was detected in the CSF specimen; however, there were no other findings suggesting infectious mononucleosis. Immunoglobulin G against the viral capsid antigen (VCA), Epstein-Barr nuclear antigen, and Epstein-Barr early antigen were all positive, but immunoglobulin M against VCA was negative. Therefore, although the diagnosis of EBV encephalitis was uncertain, acyclovir was continued for 2 weeks due to lack of an alternative diagnosis. The patient recovered fully and was discharged. No positive results were reported in the autoimmune encephalitis panel after discharge (six neuronal antibodies including anti-N-methyl-D-aspartate receptor, anti-╬▒-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, anti-dipeptidyl peptidase-like protein-6, anti-leucine-rich glioma-inactivated 1, anti-contactin-associated protein-like 2, and anti-╬│-aminobutyric acid type-B receptor antibodies). She was well, without severe headaches or seizures, until the second episode.
One and a half years later, the patient again visited the outpatient clinic because of a headache lasting 2 to 3 days. The headache was severe on the left temporal side, had a pulsating nature, and lasted almost all day. It was aggravated by movement. The headache was accompanied by nausea and vomiting, but improved after sleeping. Initially, migraine was presumed based on a family history of migraine and a normal neurological examination. However, the next day, she was admitted to the hospital because of a severe morning headache and vomiting. Subtle signal changes were observable in the temporoparietal convexity on brain MRI, but were not recognized then (Fig. 1E-H). Brain MRA was normal. The day following admission, the patient experienced severe headache with right hemiparesis, aphasia, and confusion. Neurologic deficits and consciousness completely recovered after several hours, but she had no memory of the episode. Brain MRI was reexamined using a stroke protocol. Sulcal hyperintensity in the left Sylvian fissure and temporoparietal convexity with prominent enhancement were noted on the T2-FLAIR sequence (Fig. 1I-M). Cortical hyperintensity was also observed, but was less evident than the sulcal hyperintensity. Subtle diffusion restriction was observed in the corresponding regions. Although perfusion decreased in the involved area, MRA was normal. Neither CSF pleocytosis nor fever was observed. The headache spontaneously improved, and no pathogens were detected in the CSF. Thus, flunarizine was prescribed to address the potential hemiplegic migraine, and she was discharged on day 6 after admission. After discharge, she was symptom-free for 3 days, but visited the hospital again with severe headache, vomiting, and irritability. This time, brain MRI revealed a slight increase in cortical hyperintensity; however, the enhancement was weakened. Perfusion increased in corresponding regions (Fig. 1N-R). CSF pleocytosis (WBC count, 21/mm3; lymphocytes, 85%) was observed. High-dose steroids (30 mg/kg/day for 3 days) were administered as autoimmune encephalitis was suspected. Polymerase chain reaction for human herpesviruses responsible for encephalitis (types 1 to 6), Gram stains, and culture results were negative using CSF. After steroid administration, the symptoms improved rapidly. Following high-dose steroid treatment, 2 mg/kg/day was used for 3 days and 1 mg/kg/day was used for 3 days. After that, the dose was reduced by 5 mg every 3 days for 30 days. On day 7 of steroid treatment, the anti-MOG antibody was reported to be positive (the level was 0.95 by a fluorescence-activated cell sorting cell-based array using the ratio of positive cells for analysis; >0.254 was considered as a positive result). She was discharged without sequelae and did not experience recurrence for 6 months. Fig. 2 summarizes the patientŌĆÖs clinical course.
The two distinct episodes allowed us to diagnose anti-MOG-associated encephalitis, usually classified as a sub-phenotype of CCE. During the first episode, the autoimmune encephalitis panel was examined, but MOG antibodies were not included in this panel. The MOG antibodies were detected during the second episode; however, the autoimmune encephalitis panel was not tested. There may be disagreement on the diagnosis of anti-MOG-associated encephalitis because of the limited neuro-immunological work-up. However, the clinical presentation of the patient was consistent with those of previously reported anti-MOG-associated encephalitis cases in children [7]. Fever, headache, seizures, focal neurologic deficits including motor weakness, and altered mental state are the main symptoms of anti-MOG-associated encephalitis; the patient had all these symptoms except fever, which was described as occurring in 50%-78% cases of anti-MOG-associated encephalitis [6,7]. The MRI findings of our patient were consistent with those of anti-MOG-associated unilateral CCE reported by Ogawa et al. [2]. Sulcal and cortical hyperintensities were observed in T2-FLAIR sequences during both episodes, even though the region involved seemed to shift. During the second episode, contrast enhancement of the leptomeninges was clearly demonstrated. This is suggestive of the present case, which also demonstrates a previously reported pathological finding in anti-MOG-associated encephalitisŌĆöspecifically, lymphocyte infiltration of the subarachnoid space [8]. Furthermore, serial brain MRI examinations during the second episode revealed important clinical implications. Leptomeningeal inflammation was likely to be the initial pathology because sulcal hyperintensity and enhancement were prominent at first, whereas cortical hyperintensity became evident later.
MOG antibody testing is infrequently included in the diagnostic work-up of patients with suspected autoimmune encephalitis [6]. A recent prospective multicenter observational study investigating cohorts with acute demyelinating syndrome and encephalitis revealed that anti-MOG antibodies were the most common autoantibodies, surpassing all neuronal antibodies combined in the encephalitis cohort [6]. The researchers of that study underscored that many cases of anti-MOG-associated encephalitis would have been misdiagnosed if not for the prospective study design. Our case strengthens this observation. The first episode of the present case was initially diagnosed as EBV encephalitis due to lymphocyte-predominant-pleocytosis coupled with EBV detection in CSF and findings suggestive of leptomeningeal inflammation. However, since the patient presented with a headache that progressed over 2 weeks without fever, an autoimmune condition rather than an acute viral infection of the CNS should have been suspected. Our case suggests that anti-MOG antibodies should be included in the evaluation of encephalitis with an unusual presentation, even if a pathogen is detected.
The second episode was considered a hemiplegic migraine until the anti-MOG antibody was detected. The symptoms did meet the diagnostic criteria for hemiplegic migraine, including severe pulsating unilateral headache, transient paresis, and aphasia. Furthermore, hypoperfusion on the contralateral side of the hemiparesis was also suggestive of a hemiplegic migraine [9]. However, we needed to consider the MRI findings together, including the observed sulcal hyperintensity and enhancement. Matoba et al. [10] also reported a case of anti-MOG-associated CCE mimicking hemiplegic migraine. These findings suggest that some patients diagnosed with hemiplegic migraine may have anti-MOG antibodies. Hypoperfusion observed immediately after hemiparesis could be a post-ictal change, considering the hyperperfusion seen after paresis recovery.
The present case emphasizes that anti-MOG antibodies should be included in the diagnostic process for unexplained migraine-like headaches, as well as encephalitis.
This study was approved by the Institutional Review Board of Gyeongsang National University of Hospital, Jinju, Korea (IRB No. GNUH 2022-08-015). Informed consent was waived by the board.