Cerebroretinal microangiopathy with calcifications and cysts-1 (CRMCC1), also known as Coats plus syndrome, is an autosomal recessive, multisystem disorder characterized by obliterative angiopathy of small vessels, primarily in the brain, eyes, bone, and gastrointestinal tract. The clinical features of this condition include prenatal and postnatal growth restriction, bilateral retinal telangiectasias and exudates, intracranial calcification, leukoencephalopathy sometimes associated with parenchymal cysts, osteopenia with a tendency for fracture, bone marrow suppression, and gastrointestinal bleeding with cirrhosis. Less frequently, patients demonstrate sparse, gray hair, dystrophic nails, and caf├® au lait patches [1]. It has no known effective treatment to date. Bevacizumab, a vascular endothelial growth factor (VEGF) inhibitor, has been successfully used in the treatment of two disorders that share similar pathologies with a more limited distribution: Coats disease and leukoencephalopathy with calcifications and cysts (LCC), also known as Labrune syndrome [2,3]. We report the case of a patient who was diagnosed with Coats plus syndrome and demonstrated almost complete freedom from seizures, control of severe gastrointestinal tract bleeding, and a noticeable improvement in motor function, dystonia, and brain imaging findings on bevacizumab.
The male proband was born at an estimated gestational age of 26 weeks via cesarean section due to decreased fetal movement, with a birth weight of 789 g. The Apgar scores were 4 and 6 at 1 and 5 minutes, respectively. Initial head ultrasonography showed right and left germinal matrix hemorrhages without ventriculomegaly, which resolved after around 7 weeks. Term-equivalent brain magnetic resonance imaging (MRI) showed foci of bilateral germinal matrix hemorrhage and right posterior intraventricular hemorrhage, but was otherwise unremarkable.
Initially after leaving the neonatal intensive care unit, the patient had normal development. At his follow-up appointment with the newborn medicine clinic at the age of 9 months (5.5 months corrected), he screened normal on the Denver II developmental screening test. Concerns about development were raised around 15 to 18 months of life and were initially attributed to cerebral palsy. He spoke his first words at 16 months (12.5 months corrected) and walked at 18 months of age (14.5 months corrected). During that period, he was noted to have ataxia and spastic diplegia, which progressively worsened, with asymmetric dystonia affecting the right side of the body more than the left. Initially, he was able to walk independently but used a walker for long distances and required some assistance with activities of daily living. He developed generalized tonic-clonic seizures around 2 to 3 years of age, which were controlled with anti-seizure medications. He also had poor visual acuity (20/125 in the right eye and counting fingers at 3 feet in the left eye), which was partially related to retinopathy of prematurity (ROP). However, due to the eye operations he underwent for ROP and resultant retinal scarring, an evaluation for the typical eye findings associated with Coats plus syndrome was not possible.
A computed tomography scan performed after the patient fell and hit his head at the age of 3 years revealed extensive intracranial calcifications, prompting brain MRI that showed multiple cystic lesions as well as hyperintensities on the T2 fluid-attenuated inversion recovery (FLAIR) sequence suggestive of white matter disease. After extensive metabolic genetic tests yielded uninformative results, he underwent clinical trio exome sequencing, performed by GeneDx, which revealed two pathogenic variants in CTC1. The Agilent Clinical Research Exome kit was used to target the exonic regions and flanking splice junctions of the genome. The targeted regions were sequenced using an Illumina HiSeq sequencing system with 100 base pairs paired-end reads. The variants were confirmed by Sanger sequencing. Variant annotation and analyses were performed using the companyŌĆÖs custom-developed analysis tool. Variant nomenclature was based on the transcript NM_025099.5, and variant classification was conducted according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines [4]. The first variant was paternally inherited and pathogenic, designated as c.724_727delAAAG;p.Lys242Leufs*41, which has been previously seen in the context of CRMCC [5] and reported four times as a pathogenic variant in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/RCV000023986/). The second variant was maternally inherited and likely pathogenic, designated as c.1459A>G;p.Arg487Gly. This variant has not been previously reported among patients with CRMCC, and it has been listed in ClinVar once as pathogenic and twice as a variant of uncertain significance (https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000392201.5). These results confirmed the diagnosis of Coats plus syndrome.
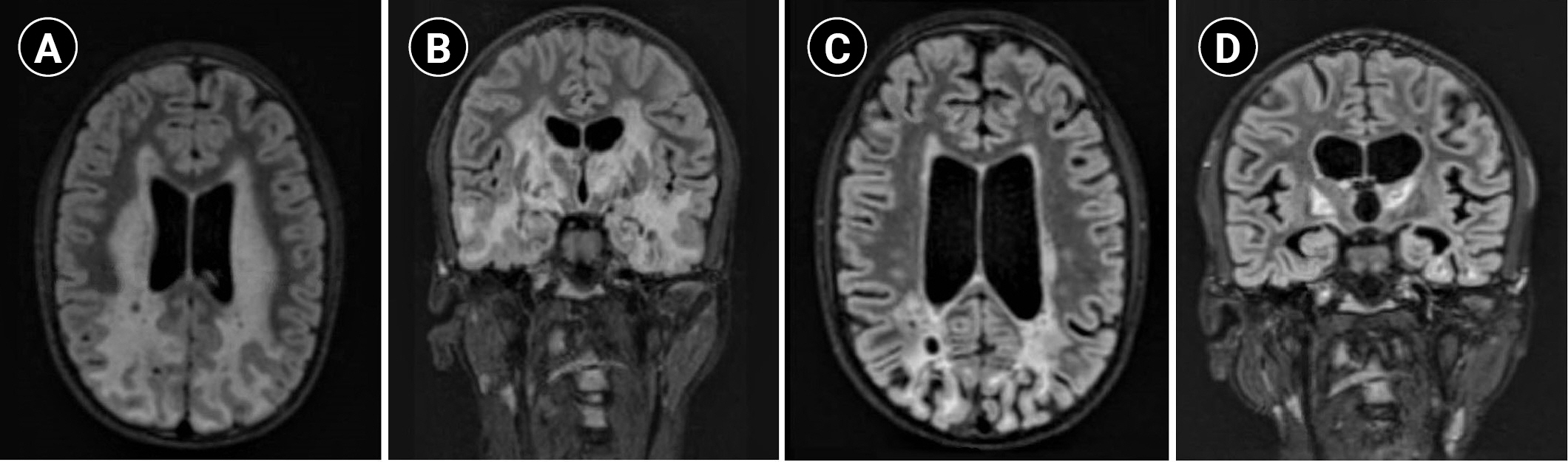
Between the ages of 11 and 15, the patient developed dysarthria that progressed to complete mutism, worsening dystonia (involving the trunk and limbs), difficulty swallowing, increasing seizure frequency (monthly seizure clusters despite continuous up-titrating of the anti-seizure medication he was on, clobazam), and episodes of severe gastrointestinal bleeding necessitating erythrocyte transfusion. He also started to experience pathological bone fractures, mainly involving the lower limbs, which affected his ability to ambulate independently and were complicated by recurrent infections, leading to the amputation of his right leg. He became wheelchair-bound but was able to use both hands to reach for objects, favoring the left side, with significant dysmetria. Danazol, an agent that has shown benefit in certain telomeropathies [6], was tried for a year and a half without any benefit. His brain MRI showed extensive cerebral, brainstem, and cerebellar T2/FLAIR signal abnormalities, cystic changes with peripheral enhancement, and parenchymal calcifications (Fig. 1A and B).
A recent case report showed improved bradykinesia, range of motion, and brain MRI findings with VEGF inhibition (bevacizumab, 5 mg/kg intravenous [IV] biweekly) in a patient with LCC [3], a disorder that overlaps clinically with Coats plus syndrome. In another report on a patient with LCC, bevacizumab treatment was associated with improvement in neuroimaging findings, accompanied by stabilization in clinical status [7]. Correspondingly, bevacizumab was started at the age of 16 (5 mg/kg IV over 60 to 90 minutes, every 2 weeks) and resulted in better seizure control (one or two clinical seizures per year), improvement in his swallowing and dystonia (with an increased range of motion in the upper extremities) and cessation of gastrointestinal tract bleeding. Brain MRI (Fig. 1C and D) obtained 4 months after the initiation of bevacizumab showed significant improvement in white matter changes. However, he developed new-onset abdominal cramps, which were thought to be related to the bevacizumab and persisted despite dose reduction (to 2 mg/kg IV, every 2 weeks). As a result, bevacizumab was stopped after 7 months and he was switched to sirolimus, an mTOR signaling inhibitor that has been shown to be effective in the treatment of vascular anomalies [8]. However, the patient experienced progressive gastrointestinal bleeding while on sirolimus; thus, bevacizumab was resumed at the same initial dose with analgesics, which successfully controlled the bleeding for 2 months before it worsened again, despite the addition of octreotide. Of note, his brain MRI remained stable during that time, but after 3 months of recurrent gastrointestinal bleeding secondary to liver cirrhosis and multiple transfusions, he was transitioned to hospice care and passed away at the age of 16 years and 9 months.
Coats plus syndrome is a rare, autosomal recessive disorder caused by mutations in CTC1, encoding conserved telomere maintenance component 1, a member of the mammalian homolog of the yeast heterotrimeric CST (Ctc1, Stn1, Ten1) telomeric capping complex [5]. A pathological examination of the organs involved suggests that the underlying basis is obliterative microangiopathy, with vessels showing thickened, sclerotic, and calcified walls and telangiectasia [1]. Coats disease, a phenotypically similar but genetically distinct disorder, shares the same ocular pathology as Coats plus syndrome, which is characterized by retinal telangiectasia, exudative retinopathy, and neovascularization. The extra-neurological problems differentiate Coats plus syndrome from LCC, in which affected individuals otherwise show an identical neuroradiological appearance [9]. LCC is characterized by cerebral microangiopathy that causes progressive white matter disease, calcifications, and cysts within the brain caused by biallelic mutations in the box C/D small nucleolar RNA SNORD118 [10]. VEGF promotes neovascularization and vascular permeability, and VEGF inhibition has been reported to reduce exudates and cysts in ocular disorders such as macular edema and Coats disease [2]. Given these observations, we reasoned that VEGF inhibition might also be effective in treating Coats plus syndrome. After starting bevacizumab, our patient experienced improved attentiveness, swallowing, dystonia, and range of motion, relatively better seizure control, and cessation of gastrointestinal tract bleeding for months before the syndrome progressed again. His brain imaging showed marked improvement in white matter T2/FLAIR hyperintensities after 4 to 5 months of bevacizumab treatment. This is the first case reported where an anti-VEGF agent was used to treat neurologic manifestations of Coats plus syndrome. Further studies are warranted, and the risk-benefit analysis of this off-label drug use must be weighed on a case-by-case basis.
Informed consent was waived by the Washington University in St. Louis Institutional Review Board (IRB ID: 202207071). Written informed consent by the patient was waived due to a retrospective nature of our study.