Microcephaly is defined as a head circumference more than 3 standard deviations (SD) smaller than the typical value in babies of the same sex and age and can result in asymptomatic to severe developmental delays and refractory seizures. Unlike primary microcephaly, which is present at birth, secondary microcephaly is characterized by a normal head circumference when the infant is born and a rapid decrease in head growth thereafter. Secondary microcephaly is also referred to as postnatal microcephaly and is sometimes associated with rare genetic disorders [1]. Intellectual developmental disorder with microcephaly and pontine and cerebellar hypoplasia (MICPCH) is an X-linked disorder affecting females that is characterized by severely impaired intellectual development, microcephaly, and varying degrees of pontocerebellar hypoplasia [2]. Calcium/calmodulin-dependent serine protein kinase (CASK)-associated MICPCH is extremely rare, and more than 50 females and a few males with MICPCH have been described in the medical literature [3]. Here, we report a very rare Korean case of an infant with MICPCH with a CASK mutation. This case was reviewed and approved by the Institutional Review Board of Keimyung University Dongsan Hospital (IRB No. 2022-07-032). The requirement for informed consent was waived by the board.
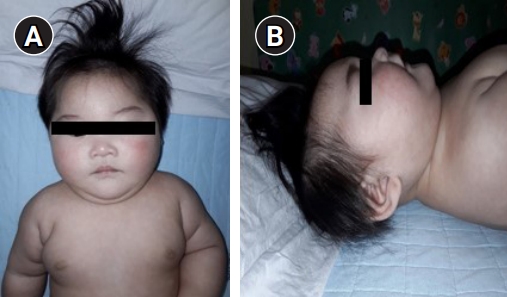
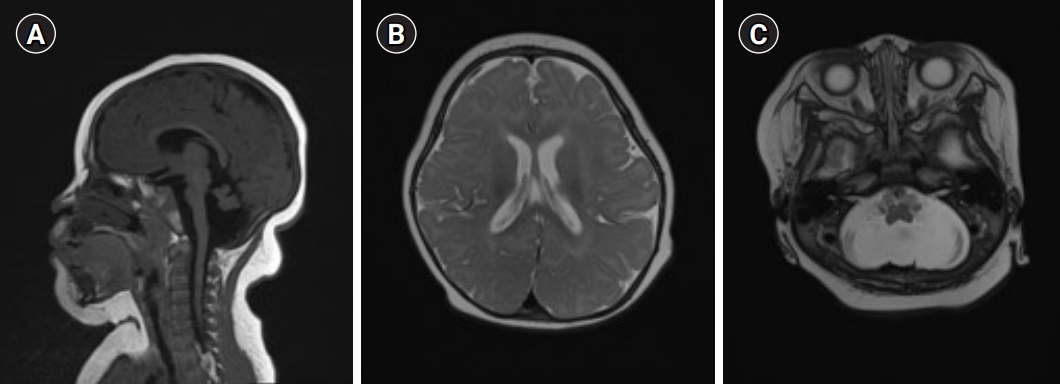
A female infant was born at 40 weeks of gestation by vaginal delivery at a local hospital. There were no remarkable complications during the pregnancy. The infant had a birth weight of 3,200 g (50th percentile), a height of 49.5 cm (50th to 75th percentile), and a head circumference of 33.5 cm (25th to 50th percentile). Immediately after birth, the auditory brainstem response test results were 35 dB on the left and 60 dB on the right and she had hearing aid treatment. She was found to have severe microcephaly (<1st percentile) persistently in routine infant check-ups (at 2 months), so she underwent brain computed tomography (CT) when she was 5 months old at another clinic. Cerebellar hypoplasia was found on the brain CT. When she came to our clinic (at 6 months), her weight was 7,830 g (50th to 75th percentile), her length was 65.3 cm (25th to 50th percentile), and her head circumference was 37.7 cm (-4 SD [38.1 cm]). She had facial particularities, including an oval face, micrognathia, arched eyebrows, hypertelorism, long eyelashes, epicanthus, low-set and prominent ears, long philtrum, and a short nose with a broad nasal bridge and tip (Fig. 1). A small anterior fontanelle was found in the physical examination. The physical examination also found increased muscle tone in the lower extremities. Brain magnetic resonance imaging (MRI) findings showed microcephaly with diffuse smooth gyral markings of the cerebral cortex, and pontocerebellar hypoplasia (Fig. 2). Congenital viral and metabolic workup test results were all normal. Her family history was not significant, and her parents were healthy and denied consanguinity.
Conventional chromosomal analysis revealed a normal female karyotype (46, XX). In next-generation sequencing (hereditary microcephaly panel), the heterozygote c.2400C>G (p.Tyr800Ter) variant was found in the CASK gene. The CASK gene is the causative gene of intellectual developmental disorder and MICPCH (OMIM #300749), which is inherited in an X-linked dominant manner and is characterized by severely impaired intellectual development and varying degrees of pontocerebellar hypoplasia. The patientŌĆÖs features and symptoms were consistent with those of MICPCH. CASK c.2400C>G is a novel variant that has not been reported in previous publications or in normal population databases, including gnomAD/ExAC (https://gnomad.broadinstitute.org/), the 1000 Genomes Project (http://1000genomes.org), and the Korean Reference Genome Database (http://coda.nih.go.kr/coda/KRGDB/) (PM2). Loss-of-function is a well-known mechanism of disease associated with the CASK gene, and c.2400C>G (p.Tyr800Ter) is a null variant expected to cause nonsense-mediated decay (PVS1). Whether this variant occurred de novo or was inherited in this case could not be determined because the parents refused genetic testing. However, neither parent showed any specific symptoms or abnormal findings, and the penetrance of MICPCH is known to be complete, so the probability of a de novo variant is very high. According to the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines, we applied PVS1 and PM1 as evidence of pathogenicity and interpreted this to be a likely pathogenic variant. The patient had no seizures and showed developmental delays at 6 months. All developmental domains were at the level of 3 to 4 months. She is undergoing continuous rehabilitation treatment and follow-up.
CASK-related disorders have two main clinical phenotypes, including (1) MICPCH, which primarily affects females and generally is associated with pathologic loss-of-function variants in the CASK gene and (2) X-linked intellectual disability with or without nystagmus has been reported in males and females, and is generally associated with hypomorphic CASK pathogenic variants such as missense variants and in-frame deletions [4]. Mental retardation and MICPCH are diagnosed in females with severe intellectual disabilities and progressive microcephaly, and usually include epileptic seizures [4]. The head circumferences initially range from normal to mild microcephaly (approximately -2 SD), followed by mild to severe progressive postnatal microcephaly (less than -10 SD) around 4 months of age [5]. Patients have characteristic facial malformations (broad forehead, oval face, arched eyebrows, hypertelorism, epicanthus, macrotia, long philtrum, short nose with a broad bridge, large eyes, and micrognathia) and ophthalmic (nystagmus, optic nerve hypoplasia, glaucoma, and myopia) and auditory (hearing loss in about 25% of cases) manifestations [2,5]. Neurological symptoms such as seizures (about 40% of the patients), hypotonia, extremity hypertonia, or dystonia may also occur [4]. Only about 25% of the patients can walk, and most of them do not develop language for communication [3,6]. The patient in our case also showed severe microcephaly after birth, and a broad forehead, small chin, arched eyebrows, long eyelashes, large ears, long philtrum, broad nose bridge, and short nose with tip were observed, which are findings consistent with MICPCH. In this case, too, there was hearing loss immediately after birth, which is thought to be a symptom of MICPCH. To date, more than 70 CASK mutations have been identified and reported, and CASK mutations have been shown to manifest as a wide range of phenotypes [7]. However, the disease has several common features, including severe developmental delay, severe postnatal microcephaly under 1 year of age, and with or without extremity hypertonia and postnatal growth retardation [7].
The CASK gene is located at Xp11.4 and is a member of the membrane-associated guanylate kinase protein family. CASK is believed to manipulate the formation and activity of synapses by controlling presynaptic tissue and neurotransmitter release. In addition, CASK can modulate gene expression involved in cortical development, maintain dendrite morphology, and control ion channels to their post-synaptic locations [8]. Due to the location of CASK on the X chromosome, MICPCH usually results from loss-of-function mutations and is overrepresented in females due to early male mortality [9].
The MRI findings of mid-hind brain hypoplasia and normal or large corpus callosum in girls with microcephaly and neurodevelopmental retardation may suggest a possible CASK mutation [2,10]. In brain MRI examinations of patients with CASK mutations, the supratentorial abnormalities included delayed myelination and progressive cortical atrophy, simple gyrus, subcortical high T2 signal abnormality in the frontal cortex, and venous hemangioma [2-4]. The MRI findings in our case showed the same results as in other studies, and normal corpus callosum and diffuse smooth gyral markings of the cerebral cortex were also observed.
To date, fewer than 100 cases of MICPCH have been reported, making it a very rare disease. Our patient presented a previously unreported mutation in the CASK gene consistent with the MICPCH phenotype. We believe that the description of this case may be useful in expanding our knowledge of MICPCH syndrome.