Myoclonic epilepsy of infancy, myoclonic-atonic epilepsy (MAE) with onset in early childhood, and later-onset syndromes such as juvenile myoclonic epilepsy, eyelid myoclonic epilepsy, and myoclonic absence epilepsy are all examples of childhood-onset myoclonic epilepsy syndromes. Degenerative brain disorders, subacute sclerosing panencephalitis (SSPE), autoimmune disorders, and a few mitochondrial abnormalities are among the other uncommon causes. There have been attempts to define hereditary myoclonic epilepsies that do not meet the recognised criteria for myoclonic epilepsy syndromes [1]. The cognitive outcomes of epilepsy syndromes vary, but in general, they have a good prognosis. Myoclonic-atonic epilepsy is classified as an epileptic encephalopathy, and the cognitive outcome can be normal to severely impaired [2].
SSPE, in contrast, is a debilitating neurological illness that has so far eluded treatment. It is characterised by cognitive decline, periodic myoclonus, ataxia, vision complaints, a vegetative state, and death [3]. Clinicians usually identify SSPE using a combination of classic clinical features paired with elevated anti-measles antibody titres in the cerebrospinal fluid (CSF). Thus, a problem arises when a clinician is faced with a case of SSPE presenting in its early stages or with atypical symptoms. In the case described herein, the initial presentation suggested SSPE with an atypical presentation, but the work-up to rule out other possibilities and careful monitoring led to a final diagnosis of MAE.
A 6-year-old boy with normal development, had repeated brief generalised tonic-clonic seizures (GTCS) for the past 4 months (1 to 2 episodes/week). Valproic acid was started at 20 mg/kg/day, but discontinued 2 weeks later due to new-onset behavioural issues and transaminitis. Levetiracetam was then initiated at 20 mg/kg/day and optimised to 25 mg/kg/day. According to the mother’s account, the seizure frequency increased to 3 to 4 episodes/week. At this point, lacosamide was started at 2 mg/kg/day and increased weekly to 4 mg/kg/day. He presented to the emergency with head drop. He had not experienced any episode of GTCS for the last one and half months. Head drop occurred more frequently in the morning. He also had progressive cognitive decline and behavioural issues such as hyperactivity, temper tantrums, irritability, and emotional lability. The child had been vaccinated against measles at 9 and 15 months of age, and had never had clinical measles. The Childhood Behaviour Checklist showed borderline scores for inattention (68) and hyperactivity (67). He had no dysmorphism or neurocutaneous markers, and the neurological examination was essentially normal. The eye examination revealed no pigmentary, necrotising, or optic atrophy, and the hearing test was normal. Autoimmune encephalitis, SSPE, and progressive myoclonic epilepsies were the first clinical possibilities. His blood count, arterial lactate, ammonia, glucose, and ketone levels were in the normal range. Human immunodeficiency virus (HIV) serology was negative. The CSF showed two cells/mm3 (lymphocytes), 15 mg/dL protein, and 61 mg/dL glucose (random blood sugar 96 mg/dL), sterile CSF culture, and a negative CSF viral panel. The measles antibody titres in serum and CSF determined by the enzyme-linked immunosorbent assay method are presented in Table 1.
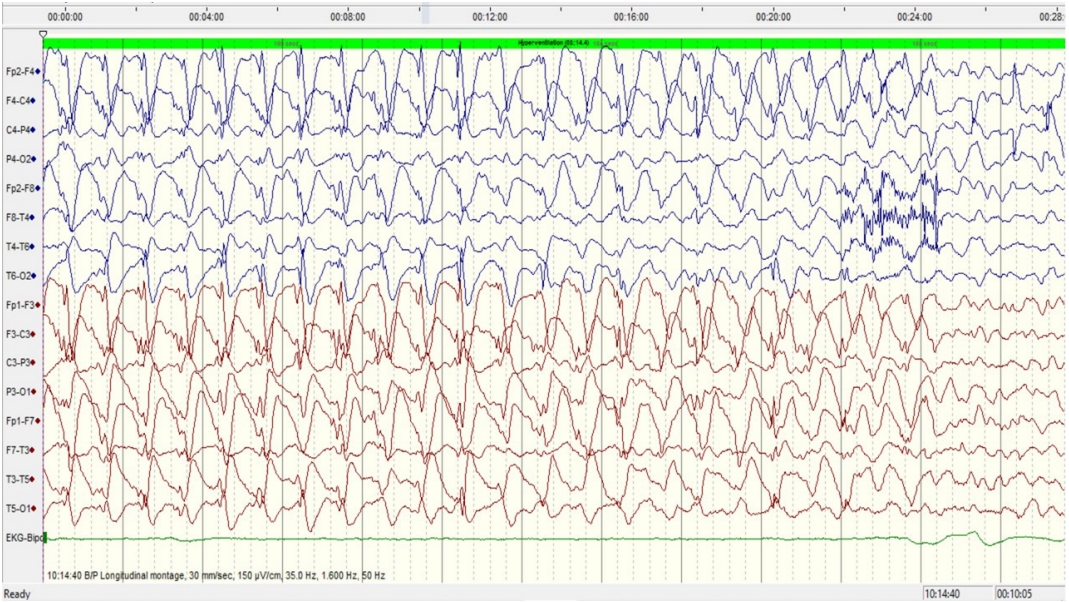
The sleep electroencephalography (EEG) record showed frequent, quasi-periodic (6 to 10 seconds) generalised bursts of spike-and-wave (2.5 to 3.0 Hz, 450 to 550 μV) and polyspike discharges lasting for 0.5 to 3 seconds (Fig. 1). Activation procedures, such as photic stimulation and hyperventilation, did not yield any additional information. Brain magnetic resonance imaging (MRI) was normal. Despite the absence of classical EEG findings, SSPE still remained a possibility due to evidence of the intrathecal production of measles antibodies.
The child was further investigated for alternative treatable aetiologies. CSF titres of anti N-methyl-D-aspartate (NMDA) receptor antibody and serum antibodies against alpha-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA), gamma-aminobutyric acid type A and B (GABA), contactin-associated protein-like 2 (CASPR2), glutamic acid decarboxylase (GAD) were negative. With a working diagnosis of seronegative autoimmune encephalitis, immunotherapy was planned. Pre-therapy, the CSF anti-measles antibody and CSF/serum ratio were obtained again from the same laboratory; the results were still positive, but with decreasing titres (CSF/serum ratio reference for measles IgG antibody, 7.7). A paired sample was also sent to two other laboratories because we continued to have doubts about the elevated anti-measles antibody titres. The child was administered intravenous immunoglobulin at 400 mg/kg/day for 5 days and intravenous methylprednisolone at 30 mg/kg/day for 5 days. The antiseizure medications were optimised (levetiracetam at 30 mg/kg/day, lacosamide at 6 mg/kg/day, clobazam at 0.5 mg/kg/day). The child was discharged and followed up after 4 weeks. The repeated CSF measles antibody titre was in the normal range. The child showed improvements in the behavioural and cognitive profile and the frequency of head drop decreased significantly. At this time, his parents complained of new paroxysms characterised by sudden cessation of ongoing activity along with a vacant stare lasting for 10 seconds, especially upon waking up. When he was made to hyperventilate for three minutes, an episode of absence seizure was noted. The same was captured in a video EEG recording that showed frequent generalised bursts of spike-and-wave discharges (2.5 to 3.5 Hz, 550 to 650 μV) lasting for 10 to 15 seconds with abrupt onset and offset and no accentuation of discharges on photic stimulation (Fig. 2). In view of this additional finding, we disregarded the diagnosis of SSPE and considered a possible diagnosis of MAE. The child was started on ethosuximide (20 mg/kg/day) and continued with other anti-seizure medications except lacosamide, which was tapered off. A genetic study was not possible due to monetary constraints. The child’s seizure control and behaviour improved significantly at follow-up. The parents provided written informed consent to report their child’s case.
The diagnosis of SSPE is catastrophic for both the child and the caregivers. An atypical presentation of SSPE often leads to misdiagnosis, especially in the early stages [4]. The disease is rare in developed countries. Currently, India is one of the countries with the highest burdens of the disease. Many Indian SSPE children are lost to follow-up in the wake of being told the prognosis of the disease. With increasing awareness, more children with SSPE with atypical presentations are being seen [5-7]. Despite the high incidence of SSPE in India, an accurate and early diagnosis remains a challenge. In the case presented herein, the combination of cognitive decline, myoclonus, periodic/quasiperiodic high amplitude epileptic discharges, and evidence of intrathecal measles antibody positivity (CSF/serum ratio, 33.8) was highly suggestive of SSPE. However, acute-onset GTCS as the type of seizure marking the beginning of illness and the absence of a history of clinical measles in a vaccinated child were the red flags that mandated a further evaluation of the child. With such a debilitating course, one of the possibilities was fulminant SSPE, which comprises approximately 10% of all cases of SSPE [8]. GTCS, focal tonic seizures, and other seizure types have been mentioned in the literature as atypical presentations of SSPE, which delay the diagnosis of SSPE until classical features appear [8-10]. Three patterns of EEG findings have been mentioned, with periodicity being common to all. In the current case, initial EEG showed quasiperiodic bursts of high-amplitude delta waves lasting for 0.5 to 3 seconds, although there was no evidence of polymorphic waves to suggest the classical “Radermecker complexes.” The initial EEG findings did not point against the diagnosis of SSPE, but subsequent EEG findings clearly did not favour SSPE. A stormy onset of myoclonic seizures with cognitive stagnation or regression is well known in children with MAE. Usually children tend to have their first seizure by 4 years but seizure onset can be as late as 6 years. The initial EEG can be normal, while generalised 2 to 5 Hz spike wave discharges appear later. These children may also have GTCS and absence seizures. This child developed GTCS first, followed by head drop, and absence seizures appeared last with classical EEG findings. Autoimmune encephalitis is the closest mimicker of SSPE, with clinical similarities and CSF measles antibody positivity [11]. This treatable disorder can masquerade as SSPE, which is progressive and fatal. Hence, this child was given immunotherapy with initial partial response. What remains unexplained in this case is the high CSF measles antibody titres. Damage to the blood-brain barrier due to any cause may lead to a false elevation of intrathecal CSF measles antibody titres. However, to the best of our knowledge, elevated CSF antibodies in MAE have not been reported in the literature. The positive serology in the current case, with decreasing titres from a laboratory and a subsequent negative report from another laboratory, combined with the electroclinical evolution of symptoms helped us to rule out SSPE. The difference in results from different laboratories can be explained either by differences in the dilution standardisation used by different laboratories (1:2 or 1:4 dilution) or a coincidental subclinical measles infection in the child. However, the findings of normal CSF biochemistry suggest that the blood-brain barrier was intact; hence, the second hypothesis is less likely as the finding of elevated CSF antibodies could not have been a simple reflection of an increased serum antibody titre. SSPE has no diagnostic brain MRI findings except posterior predominant white matter signal changes [12].
As this case illustrates, the diagnosis of SSPE in a child with an atypical presentation should ideally be one of exclusion, and the child should be evaluated for treatable underlying causes. SSPE should be diagnosed only after careful consideration of the clinical course. Clinicians should not be biased towards the diagnosis of SSPE by positive serology in the absence of other strong indicators.
A child with MAE who presents in an unusual way may be misdiagnosed with SSPE. Positive measles antibody titres should be strongly supported by clinical and EEG features to confirm the diagnosis of SSPE.
This study was approved by the Institutional Review Board of Command Hospital Chandimandir (approval number: 1234/Paeds/CHWC). The written informed consent was taken from the parents.