Paramyotonia congenita (PC) is a type of Na+ channelopathy caused by mutations in the Na+ voltage-gated channel alpha subunit 4 (SCN4A) gene on chromosome 17q23, which encodes voltage-gated Na+ channels (Nav1.4) in skeletal muscles and is inherited in an autosomal dominant pattern [1,2]. These channels are integral membrane proteins that exist in most excitable cells. They are mainly responsible for rapid membrane depolarization, which is the initial phase of the action potential. Rapid inactivation after an action potential prevents repetitive excitation and maintains normal physiological excitability of skeletal muscles.
Abnormal activity of human skeletal muscle due to a dysfunctional Na+ channel α-subunit with an SCN4A mutation causes excessive excitability and leads to the activation or inactivation of the channel. Clinical phenotypes associated with SCN4A mutations include PC, type 2 hyperkalemic periodic paralysis (PP), type 2 hypokalemic PP, congenital myasthenic syndrome-16, and acetazolamide-responsive myotonia congenita [1]. Furthermore, PC is characterized by muscular myotonia without weakness, and it mainly affects the muscles of the neck, face, and upper limbs. PC typically occurs in infancy or childhood and is triggered by exposure to cold or physical activity [3].
In this paper, we report the case of a male adolescent and his three-generation family with PC caused by an SCN4A mutation. This case was reviewed and approved by the Institutional Review Board of Pusan National University Hospital (IRB No. 2205-013-114). Informed consent was obtained from all parents. Patients’ medical records and other data were anonymized to ensure confidentiality.
The proband was a 14-year-old boy with a 2-year history of episodic muscular stiffness and weakness. The patient often experienced weakness and stiffness in the lower and upper extremities. During exercise, the lower extremities were affected more than the upper extremities; however, symptoms were also observed in the hands, arms, and face. During the asymptomatic period, the patient exercised normally. However, once symptoms developed, the patient was unable to move. When exposed to cold environments, such as when washing the face or during cold weather, symptoms were aggravated in the exposed body parts.
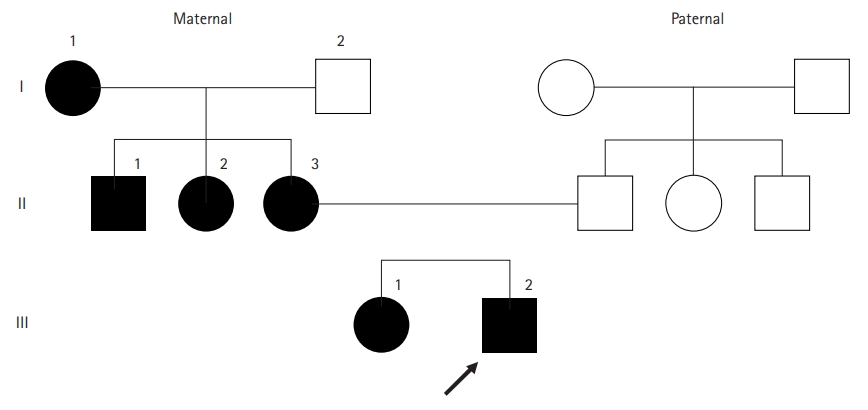
The patient was born at a gestational age of 40 weeks via normal vaginal delivery, weighing 3,400 g. He showed normal development and growth. The mother of the patient also had stiffening and muscle weakness in the upper extremities, face, and lower extremities from around the age of 10, which worsened with cold exposure and exercise. The 16-year-old sister of the patient also showed similar symptoms from around the age of 9. The patient’s maternal aunt and grandmother presented similar symptoms. All laboratory tests, including complete blood count, electrolytes, liver function tests, creatine kinase level, and thyroid function tests, were normal. Electromyography performed at another hospital revealed a diffuse myotonic discharge.
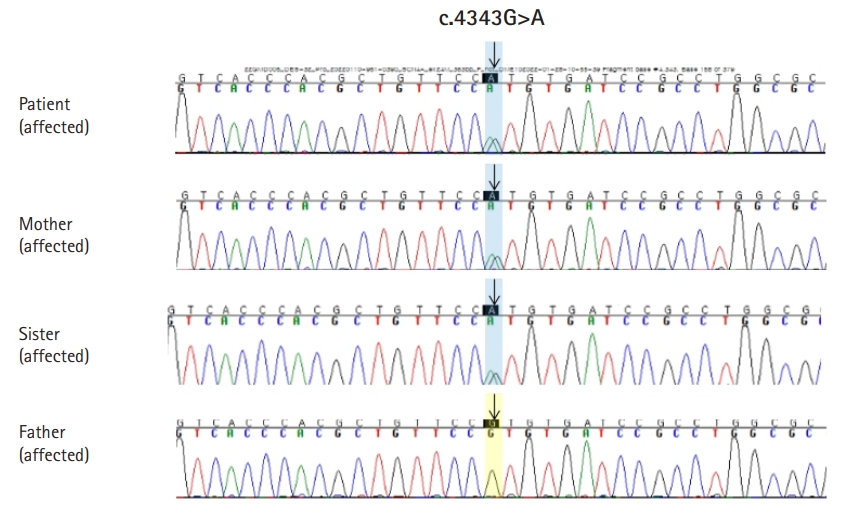
We conducted diagnostic exome sequencing (DES) of the proband. Genomic DNA was extracted from peripheral blood leukocytes and DES was performed by Green Cross Genome (Yongin, Korea) using the Celenics G-Mendeliome DES panel. The panel includes 80,962 targeted exons in 5,870 genes and used DNBSEQ-G400 (MGI Tech. Co., Shenzhen, China) with a 2×100 paired-end read method. The sequenced reads were mapped and compared with the published human genome build (UCSC GRCh37/hg19 reference). We identified a heterozygous missense mutation, c.4343G>A (p.R1448H) in SCN4A (Fig. 1). The p.R1448H variant in SCN4A has not been reported in population databases, such as gnomAD (https://gnomad.broadinstitute.org/) and Korean Reference Genome Database (KRGDB). In in silico predictions (sorting intolerant from tolerant [SIFT], polyphen, Mutation Taster), the SIFT score was 0 (deleterious), and the polyphen-2 score was 1 (probably damaging). We conducted Sanger sequencing for the variant in four family members, including the proband, a sister, and both parents. The variant was co-segregated with the affected members (proband, a sister, and a mother) within the family (Fig. 2).
The R1448H variant has been previously reported in PC patients and has been found to be related to the phenotype of PC in functional studies [4]. Thus, the variant was interpreted as a likely pathogenic variant according to the 2015 American College of Medical Genetics guideline [5]. We administered lamotrigine (3 to 3.5 mg/kg/day) to the patient and his sister. The symptoms of the lower extremities improved, although intermittent dystonia in the hands and face persisted.
It has been shown that PC worsens with exposure to cold, is relieved by warm temperature, and is aggravated by continued activity (paradoxical myotonia) [1]. Symptoms are usually noticed during infancy and present in most patients as teenagers. The progression of PC with age and muscle atrophy are not observed. Symptoms can last from a few seconds to a few minutes and, in some cases, for several days. Some patients experience worsening symptoms after consuming foods that are high in potassium. Patients with PC are instructed to avoid potassium-rich foods. Although no permanent disability remains, there are limitations in activities of daily living. The treatment of PC involves avoiding sudden exposure to very cold weather and sudden intense physical activities. Potassium-rich foods can trigger muscle stiffness, and patients should know how to manage their potassium intake. Medications that block sodium channels, such as mexiletine and lamotrigine, may reduce stiffness in PC patients [2,6].
Voltage-gated Na+ channels are integral membrane proteins that conduct Na+ through cell membranes. In excitable cells, Na+ channels are responsible for the rising phase of action potentials, known as “depolarization.” There are many subtypes of Na+ channels, and Nav1.4 is mainly present in skeletal muscles. The Na+ channels consist of large α- and accessory β-subunits. The α-subunits exhibit the core function of the channel, which conducts Na+ in a voltage-gated manner, even in the absence of β-subunits. The α-subunit is encoded by the SCN4A gene at chromosome 17q23.3, and the β-subunit is encoded by the SCN1B gene at chromosome 19q13.11 [7]. Mutations of SCN4A cause several subtypes of sodium channelopathies, including PC, hyperkalemic PP, hypokalemic PP, congenital myasthenic syndrome, and atypical acetazolamide-responsive myotonia congenita [1]. Mutations of SCN1B have been associated with cardiac arrhythmias, developmental epileptic encephalopathy, and generalized epilepsy with febrile seizures [7]. Nav1.4 carries the majority of the inward Na+ current. The activation of the channel generates an action potential, and the rapid inactivation of the channel after an action potential helps the membrane potential to return to the resting potential quickly. This entire process maintains normal skeletal muscle contraction. Gain-of-function mutations in SCN4A, the gene that encodes skeletal muscle Nav1.4, enhance the inward Na+ current, which leads to increased membrane excitability and a decrease in channel inactivation [2,8]. As a result, there is an inappropriately increased excitability of action potentials due to the increased amount of available Na+ channels (impaired inactivation) and the easy opening of Na+ channels with mild depolarization (increased excitability) from resting potential due to increased extracellular Na+ levels. This pathophysiological mechanism can lead to an initial burst of myotonia discharges and results in the symptom of stiffness. SCN4A mutations produce various clinically distinct skeletal muscle disorders, including hyperkalemic PP, PC, potassium-aggravated myotonia, hypokalemic PP, and congenital myasthenic syndrome. The SCN4A gene comprises several exons and is associated with various clinical phenotypes. The clinical presentation of patients varies depending on which nucleotide, codon, or amino acid changes, and the number of mutant exons. If there is enhanced excitability with a small amount of Na+ influx, myotonia due to increased excitability can be observed. Mutations associated with prominent inactivation defects (abundant Na+ influx) with disrupted slow inactivation increase the susceptibility to PP from sustained depolarization of the resting potential [9]. After the discharge level is increased, most Na+ channels become inactive and do not respond to stimuli, resulting in paralysis symptoms [8,9].
In conclusion, PC is very rare, with an estimated prevalence of less than 1:100,000 [10]. In Korea, there have been no previous reports of PC caused by the R1448H mutation of SCN4A, and the present case is the youngest patient with PC. It is important to obtain a detailed history, recognize the characteristic symptoms of PC, and conduct appropriate diagnostic tests. We hope that this case report will enhance pediatric neurologists’ understanding of PC.