A Patient with Pyridoxine-Dependent Epilepsy Who Was Treated with Triple Therapy
Article information

Pyridoxine-dependent epilepsy (PDE) is a type of developmental and epileptic encephalopathy manifesting as seizures that are resistant to anti-seizure medication (ASM) but responsive to pharmacologic doses of pyridoxine [1]. PDE caused by bi-allelic mutations in the aldehyde dehydrogenase 7 family member A1 (ALDH7A1) gene on chromosome 5q32.2 is designated PDE-ALDH7A1 [1,2]. Mutations at this locus are associated with decreased activity of α-aminoadipic semialdehyde (α-AASA) dehydrogenase in lysine metabolism [3]. PDE-ALDH7A1 is a rare disease with an estimated incidence of 1:65,000 to 1:250,000 live births [2]. Refractory neonatal seizures are the most common presentation; however, 25% to 30% of patients were found to present with seizures outside of the neonatal period, and varying intellectual disabilities and developmental delays were found in 75% of patients [4].
We report the case of a 9-year-old boy with intractable seizures related to homozygous ALDH7A1 mutations, who improved after triple therapy. The patient had neonatal seizures on his 12th day of life. He was started on multiple ASMs, including phenobarbital, phenytoin, levetiracetam, topiramate, vigabatrin, and clonazepam; however, his seizures and the related epileptiform discharges on electroencephalography (EEG) persisted. He received empiric high-dose vitamin therapy, which included pyridoxine, inconsistently. For the next 7 years, he was admitted repeatedly for recurrent status epilepticus whenever his medications were discontinued.
When the patient was 7 years old, his father stopped giving him the prescribed vigabatrin and pyridoxine. Ten days after therapy interruption, he was admitted to the intensive care unit (ICU) for seizures, vomiting, and poor general condition. Doctors resumed vigabatrin and pyridoxine. However, the recurrence of vomiting prevented oral intake of these medications, and he had seizures again. He was readmitted to the ICU and was administered pyridoxine at 50 mg/day (2 mg/kg body weight), after which the seizures stopped.
Prompted by this clinical information, whole-exome sequencing was performed in the proband, mother, and father for an accurate diagnosis. Compound heterozygous mutations were identified in the ALDH7A1 genes: NM_001182.4:c.210C>A(p.Cys70Ter) and c.871+5G>A. The variants confirmed by Sanger sequencing were classified as pathogenic according to the guidelines of the American College of Medical Genetics and Genomics [5]. A segregation study showed both parents as carriers of the variants (Fig. 1). We diagnosed the patient with PDE and increased the pyridoxine dose to 300 mg/day (10 mg/kg). Since pyridoxine supplementation, he became seizure-free, and his EEG demonstrated improvement. However, there was no significant change in cognitive development and behavior. He was still apt to scream and run around. He exhibited violent behavior and was unable to obey commands. We initiated lysine restriction and prescribed a lysine-free amino-acid formula. We also added folic acid (1 mg/day) and arginine (4,000 mg/day; 120 mg/kg body weight). After only 4 months of lysine restriction, he was able to stay in his bed without the need for restraints and to obey his father’s commands.
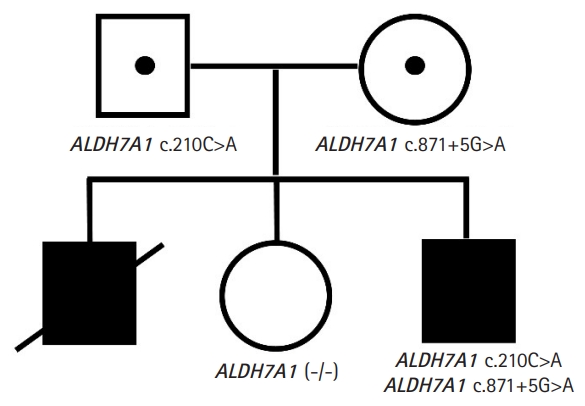
Findings of genetic testing of the patient. The patient and his relatives underwent genetic testing. Both copies of the patient’s aldehyde dehydrogenase 7 family member A1 (ALDH7A1) gene had mutations, albeit different ones. Each of these mutations was found in his father and mother, who were asymptomatic carriers. A deceased elder brother presented similarly and is assumed to have had the mutations. An elder sister showed no clinical or genetic evidence of the disease.
To diagnose PDE clinically, 100 to 500 mg of pyridoxine is administered intravenously [2] while monitoring the patient’s EEG, oxygen saturation, and vital signs [1]. Clinical seizures generally cease within several minutes in patients with PDE [1]. Caution should be exercised in administering an intravenous infusion of pyridoxine because it could cause a sudden increase in the neurotransmitter gamma-aminobutyric acid, leading to electrocerebral silence, which manifests clinically as excessive drowsiness or coma [1].
Molecular genetic testing is the only reliable method to confirm the clinical diagnosis of PDE, to diagnose the disease prenatally, and to screen family members who may be carriers [1,2]. There are no specific seizure or EEG patterns with this disease [6]; therefore, PDE needs to be tested in any cases of epilepsy with unknown etiology [2]. Additional biochemical testing for α-AASA and Δ1-piperideine-6-carboxylate in serum, urine, and cerebrospinal fluid should be performed when a single pathogenic variant or a variant of uncertain significance is identified on genetic testing [1,2].
Triple therapy consists of pyridoxine and arginine supplementation, as well as lysine restriction, and is effective in PDE-ALDH7A1 [7]. PDE-ALDH7A1 should be treated with pyridoxine supplementation of 100 mg/day in newborns, 30 mg/kg/day (maximum of 300 mg/day) in infants, and 5 to 30 mg/kg/day (maximum of 500 mg/day) in children and adolescents [2]. All patients on pyridoxine supplementation must undergo clinical screening for peripheral neuropathy (PN) [2]. Initially, only patients treated with >500 mg/day of pyridoxine were considered at risk for PN [8]. However, Ghavanini and Kimpinski [9] suggested that even pyridoxine doses as low as 50 mg/day, if used for greater than 6 months, may increase the risk of neuropathy. Lysine reduction therapies have been associated with improved long-term neurologic outcomes [2]. The use of a lysine-free formula with amino acid supplementation permits the limitation of lysine intake while ensuring adequate intake of the other amino acids [10]. Lysine is an essential amino acid and over-restriction of lysine could lead to malnutrition [8]. Thus, patients on a lysine-restricted diet should have plasma amino acids measured at least every 3 (<3 years of age) to 6 months (>3 years of age) [2]. Pharmacologic doses of arginine compete with lysine for intestinal absorption, transport across the blood-brain barrier, and entry into the mitochondria [2]. Previous recommendations for arginine dosing recommended 150 mg/kg/day if administered in addition to a lysine-restricted diet and 400 mg/kg/day if administered with pyridoxine alone [2]. Coughlin et al. [2] suggested that a higher dose of arginine (300 to 600 mg/kg/day in adults) may be required to impact lysine transport.
The patient had an elder brother who died of status epilepticus and sepsis (Fig. 1). This boy first had seizures at 2 months of age and was treated with ASMs and pyridoxine at 12.5 mg/day. When he was 7 years old, he was unable to take oral medications and supplements due to vomiting, and he eventually died. He did not undergo next-generation sequencing, but he is presumed to have had the ALDH7A1 mutations. Because the discontinuation of pyridoxine coincided with that of the ASMs, physicians failed to relate the seizures with the withdrawal of pyridoxine.
In summary, we report the case of a boy with intractable seizures related to ALDH7A1 mutations. Triple therapy controlled his seizures and improved his behavior. This case describing the diagnosis and treatment of PDE may be of interest to pediatricians and pediatric neurologists who manage this disease.
This study was approved by the Institutional Review Board of the Gangnam Severance Hospital, Yonsei University College of Medicine (3-2017-0168). The requirement for informed consent for this retrospective study was waived by the board.
Notes
Young-Mock Lee is an editorial board member of the journal, but he was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: MR, JHN, and YML. Data curation: MR, JHN, and YML. Methodology: MR, JHN, and YML. Project administration: MR, JHN, and YML. Visualization: MR, JHN, HL, and YML. Writing-original draft: MR. Writing-review & editing: MR, JHN, and YML
Acknowledgements
The authors are grateful to all staff members, doctors, and statistical consultants who were involved in this study.