Clinical Characteristics and Neurologic Outcomes of X-Linked Myotubular Myopathy
Article information

Abstract
Purpose
X-linked myotubular myopathy (XLMTM) is a rare condition of centronuclear myopathy caused by myotubularin 1 (MTM1) mutations. Patients with XLMTM show different neurodevelopmental outcomes after the neonatal period depending on age and acquired hypoxic damage. We aim to evaluate the clinical characteristics and neurodevelopmental outcomes of patients with XLMTM who were followed up at a single center. It is essential to understand the volume and conditions to prepare for being a candidate for new therapeutic strategies.
Methods
Patients diagnosed with centronuclear myopathy by muscle pathology and MTM1 mutation analysis were included. We retrospectively investigated motor milestones, communication skills, and bulbar and respiratory function in the patients. The patients were categorized into two groups: with and without hypoxic insults (HI).
Results
All 13 patients were severely affected by neonatal hypotonia and required respiratory support and a feeding tube during the neonatal period. The follow-up duration was 4.4 years (range, 0.3 to 8.9). In the non-HI group, developmental milestones were delayed but were slowly achieved. Some patients underwent training in oral feeding with thickened foods and weaning from ventilation. Patients with HI showed poor motor function catch-up and communication skills. Three deaths were associated with acute respiratory failure.
Conclusion
Patients with XLMTM without HI can survive long-term with the slow achievement of motor milestones and bulbar and respiratory function. However, hypoxic brain damage following acute respiratory failure negatively influences their developmental potential or even lead to death. Therefore, parental education for proper respiratory management is necessary, especially for young children.
Introduction
X-linked myotubular myopathy (XLMTM; OMIM 300415) is a neuromuscular disorder pathologically categorized as centronuclear myopathy [1]. MTM1 encodes myotubularin, which is implicated in the phosphatidylinositol 3-kinase pathway and is required for muscle cell growth, differentiation, and intracellular trafficking [2-4]. Newborns affected by XLMTM usually present severe hypotonia and typically require mechanical ventilators and nutritional support [5]. Although long-term survivors depend on a wheelchair, ventilator, and tube feeding, the disease course of XLMTM is relatively stable [1]. The critical roles of a multidisciplinary unit include encouragement of exercise training to maximize patients’ motor function and independence as well as safe maintenance of feeding tubes and home ventilators [6,7]. In a cross-sectional study about a natural history of patients with XLMTM, all observed deaths were associated with respiratory failure [1]. Despite a patient surviving respiratory failure, hypoxic brain damage can cause devastating neurodevelopmental outcomes.
New therapeutic strategies have recently been identified for XLMTM in children, with studies investigating gene transfer (Gene Transfer Clinical Study in X-Linked Myotubular Myopathy, ASPIRO, NCT03199469), an antisense oligonucleotide (ASO) strategy (Early Phase Human Drug Trial to Investigate Dynamin 101 (DYN101) in Patients ≥ 16 Years With Centronuclear Myopathies, Unite-CNM, NCT04033159), and tamoxifen therapy (Tamoxifen Therapy for Myotubular Myopathy, TAM4MTM, NCT04915846). To participate in clinical trials, it is essential to understand the volume and their conditions of domestic patients [8-11]. Data regarding patient characteristics must be shared to allow careful monitoring.
This study is the first to review the clinical characteristics and neurological outcomes, including motor milestones, communication skills, and bulbar and respiratory function, in Korean pediatric patients with XLMTM. To evaluate the neurological consequences of hypoxic events, we described the neurological status of patients with XLMTM divided into the following two subgroups: the hypoxic insults (HI) group and the non-HI group.
Materials and Methods
1. Patients and genetic studies
Male patients with a confirmed muscle biopsy result consistent with centronuclear myopathy and an MTM1 mutation from January 2004 to August 2021 were included. MTM1 mutations were identified by a single-gene sequencing or next-generation sequencing (NGS) panel of congenital myopathy-related genes. For positive probands, maternal segregation analyses were performed. The medical records were retrospectively reviewed, including the genotype, prenatal/postnatal history, clinical features of XLMTM, developmental milestones in motor and language, dependency on a ventilator and feeding tube from the neonatal stage to childhood, and mortality. We compared neurodevelopmental outcomes between the HI and non-HI groups. The Institutional Review Board (IRB) of the Seoul National University Hospital approved this study (IRB no. 1101-110-353) and waived the requirement for informed consent.
Results
1. Clinical features
The median age of the 13 patients was 4.4 years (range, 0.3 to 8.9). Muscle biopsies were performed at a median age of 3.7 months (range, 0.9 to 11.3). All patients presented with neonatal hypotonia (Table 1). Among them, preterm birth (before 37+0 gestational weeks) occurred in nine patients (69.2%), and asphyxia at birth was reported in five patients (38.5%). All patients received nutritional and respiratory support after birth. Respiratory support consisted of intermittent positive-pressure ventilation/synchronized intermittent mechanical ventilator (n=7, 53.8%), use of continuous positive airway pressure/bilevel positive airway pressure (BiPAP) (n=3, 23.1%), and oxygen supply (n=2, 15.4%). The accompanying clinical features were myopathic face (n=7, 53.8%), facial weakness or ophthalmoplegia (n=5, 38.5%), a high-arched palate (n=5, 38.5%), club foot/joint contracture (n=3, 23.1%), pigeon/funnel chest (n=5, 38.5%), and undescended testis (n=10, 76.9%).
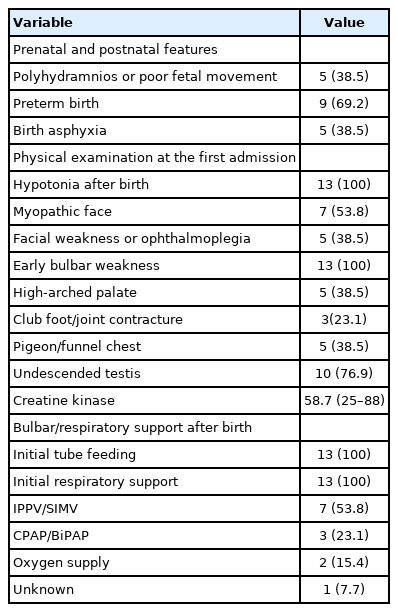
Clinical features of patients with XLMTM (n=13)
2. Genotype
Among the 13 patients, MTM1 mutation was identified in 11 patients (84.6%) by single-gene sequencing and in two patients (15.4%) by a NGS panel. Maternal inheritance was revealed in nine patients (69.2%) (Table 2). We identified 11 pathogenic MTM1 variants, including four splice-site mutations, three missense mutations, two deletions, one frameshift mutation, and one nonsense mutation. Three patients (no. 7, no. 9, and no. 11) had the same variant, c.342_342+4del (NM_000252.3).
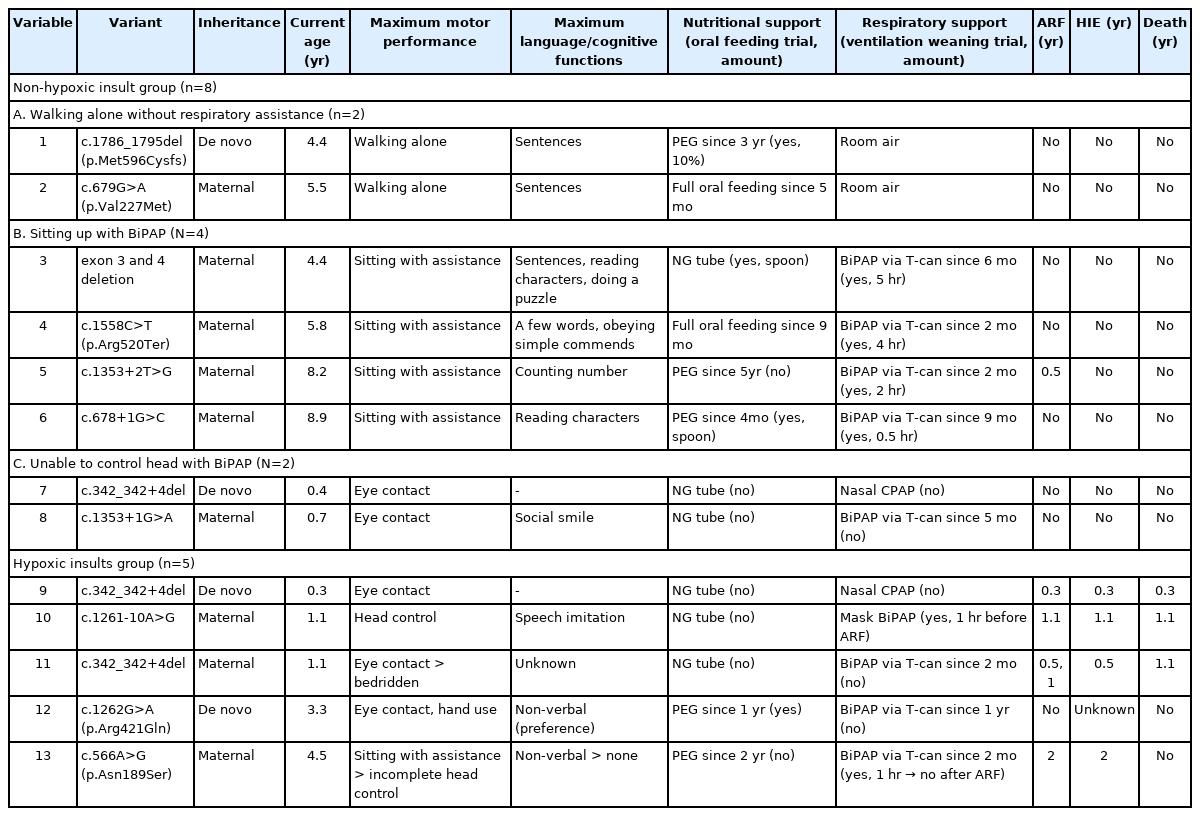
Neurodevelopmental outcomes in patients with XLMTM
3. Neurological outcomes
1) Motor and language development
Eight patients (61.5%) in the non-HI group and five (38.5%) in the HI group had different neurodevelopmental outcomes in terms of maximum performance of motor and language function (Table 2). The presented results include the neurological status at the last outpatient visit. No patient showed loss of motor and language functions. In the non-HI group (median follow-up duration, 5.0 years [range, 0.4 to 8.9]), patients slowly achieved motor milestones such as sitting up without assistance and walking alone. Verbal communication was compatible with their age. Three instances of mortality occurred due to acute respiratory failure in the HI group (median follow-up duration, 1.1 years [range, 0.3 to 4.5]). One survivor (no. 13) became bedridden after resuscitation at 2 years of age. Patient no. 12 showed severe developmental delay and HI, which was revealed by brain magnetic resonance imaging, without any prior hypoxic event. The patient showed eye contact, minimal hand use, and non-verbal communication at the age of 4.5 years.
2) Bulbar dysfunction
All patients received nutritional support through a nasogastric tube after birth. Percutaneous endoscopic gastrostomy (PEG) was performed on five patients (38.5%) at a median age of 2.4 years (range, 0.3 to 4.6). According to a videofluoroscopic swallow study, some patients underwent a challenge with oral feeding with thickened food. Two patients in the non-HI group were able to switch to full oral feeding. Both survivors in the HI group underwent PEG after revealing hypoxic brain insults.
3) Respiratory dysfunction
Eight patients (61.5%) underwent tracheostomy at a median age of 3.8 months (range, 1.6 to 11.5) and in them prolonged intubation was maintained due to failure of ventilator weaning and airway protection from respiratory emergencies was required [12]. In the non-HI group, two patients were discharged without respiratory support on room air. Four patients underwent trained ventilation weaning (weaning time range, 0.5 to 4 hours) through respiratory rehabilitation, but the ventilator support was provided for >12 hours per day.
Discussion
This study reported the neurodevelopmental outcomes, including motor function, language skills, and swallowing and respiratory function in patients with XLMTM who were followed up at a single center. Similar to our results (age range, 0.4 to 8.9 years) in a prospective study of 45 patients with XLMTM, patients <10 years (age range, 3.5 months to 56.8 years) presented slow improvements in objective muscle functions [2]. However, older patients with ventilation support for >12 hours per day showed accelerated loss of motor function. Because the aforementioned study covered a wide variation in patient ages, the measurement tools for muscle strength and motor function had been designed to accommodate very weak patients. These tools included grip and pinch strength tests, the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders scale for patients <2 years of age, the Motor Function Measure scale for patients >2 years of age, the MoviPlate device for upper limb motor function testing, and the North Star Ambulatory Assessment scale for ambulant patients. Pulmonary function tests showed results below the normal range, even in patients without ventilator support. In our study, six patients in the non-HI group required ventilator support for >12 hours per day; therefore, a relatively earlier loss of motor function would be expected in those patients than in the two patients without ventilator support (Table 2).
In our cohort, the leading causes of respiratory failure included T-cannula obstruction, T-cannula omission, or aspiration pneumonia. Because patients with XLMTM are at risk for acute respiratory failure, proper education and techniques, such as chest compression, airway clearance by tracheal suction, T-cannula obstruction response, and Ambu bag ventilation, are essential for the management of respiratory emergencies. Emergencies are more likely to occur at a younger age; thus, close attention is required [13]. Repeated training with time intervals might be helpful.
Genotype-phenotype studies have shown that most pathogenic MTM1 variants, regardless of the mutation type, resulted in loss-of-function effects, leading to the classic and severe phenotype [1,14,15]. Our cohort included all reported pathogenic variants, which were identified as three missense mutations and 10 loss-of-function mutations, including deletion, nonsense, frameshift, and splice-site mutations [16,17]. Additionally, a phosphatase domain in exon 11 is critical for maintaining protein function, but our cases did not include a variant located in the phosphatase domain [14]. In the non-HI group, only one patient (no. 2) with a missense mutation (c.679G>A [p.Val227Met]) walked alone without respiratory assistance, consistent with a mild phenotype. Although patient no. 1 had a frameshift mutation, he presented a mild phenotype. Because the frameshift mutation was located at the end of the MTM1 gene (exon 15), it would be expected to produce a relatively stable protein. All patients assisted with BiPAP had loss-of-function mutations, and the duration of BiPAP support differed among patients. Because patients no. 7 and no. 8 were <1 year of age, the serial follow-up will reveal their maximum motor achievement and amount of ventilator support. The HI group had two missense and two splice-site mutations, and the accordance data did not associate survival with these mutation types [1,14].
In the non-HI group, swallowing and self-respiratory function gradually improved. In two patients, the transition to a complete oral diet was made relatively early, at 5 and 9 months. Some patients gradually tried oral intake in small amounts in childhood. Similarly, except for two patients discharged without a ventilator in the neonatal period, other patients were stable only for a few hours on room air without respiratory support. Complete recovery of swallowing and self-respiratory function was particularly difficult. Nevertheless, even a small amount of oral challenge and attempt at short spontaneous breathing without a ventilator yielded positive effects. The challenges were aimed not at achieving complete normal function but at improving the patients’ quality of life by being able to tasting food and broadening the scope of daily activity. Therefore, it is crucial to encourage rehabilitation for patients.
Multidisciplinary therapeutic approaches are emphasized, including those related to neurology, neonatology, pulmonology, gastroenterology, rehabilitation medicine, and orthopedic surgery [6,7,18,19]. The decision to conduct a diagnostic workup involving a muscle biopsy for infants with hypotonia is sometimes difficult owing to a few factors, including the general anesthesia required for invasive procedures, the risk of respiratory complications, and occasional non-specific results. Except in cases of spinal muscular atrophy, which is routinely diagnosed through genetic investigations without the need for histopathological results, the roles of a muscle biopsy are to classify the specific disease category, help clinicians choose a genetic test, and/or modify a previous diagnosis [20]. Patients with congenital myopathy present a higher concordance rate between biopsy and genetic findings than those with congenital muscular dystrophies and metabolic myopathies. Although the genetic era of neuromuscular diseases has conspicuously developed, muscle biopsy remains a valuable tool guided by rational diagnostic algorithms. Regarding treatment, clinicians should provide proper management during serial follow-up, including routine assessments of skeletal and respiratory muscle function, speech therapy for pronunciation, and the application of devices for independent walking and scoliosis depending on the patients’ ages.
There have been recent moves towards therapeutic strategies [14]. Medical approaches suggested from Mtm1 mull mouce models include phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta (PIK3C2b) inhibition and mammalian target of rapamycin (mTOR) modulation [21,22]. Moreover, in light of proven efficacy in animal models, several clinical trials of gene therapy have been initiated, including gene replacement therapy (ASPIRO, NCT03199469) and ASO-based gene knockdown (Unite-CNM, NCT04033159). The gene transfer study (ASPIRO, NCT03199469) included 26 patients <6 years of age who required mechanical ventilator support. A clinical study of the ASO-based RNA knockdown (Unite-CNM, NCT04033159) is recruiting patients >15 years of age with identified MTM1 or dynamin 2 (DNM2) mutations. The study also has an upcoming plan for children 2 to 16 years of age. Another study, focused on tamoxifen therapy for XLMTM patients (TAM4MTM, NCT04915846), is recruiting patients >1 year of age. Tamoxifen is expected to reduce DNM2 expression, resulting in changes in the triad structure and improvement of muscle contractility [23,24]. To facilitate enrollment in upcoming clinical trials, it is important to collect patients in Korea and evaluate their conditions.
However, this study had several limitations. First, it did not have a substantial degree of variation in age distribution due to the small size of the cohort. Second, data on developmental milestones were retrospectively collected from the descriptive medical records. Third, for an accurate evaluation of motor function improvement and deterioration, it is necessary to adopt measurement scales suitable for each patient’s age.
Patients with XLMTM without HI can be long-term survivors with the slow achievement of motor milestones and bulbar and respiratory function. However, hypoxic brain insults following acute respiratory failure are significant events that negatively influence the developmental potential or even lead to death. Therefore, parental education for proper respiratory management is necessary, especially when children are at a young age.
Notes
Anna Cho, Ki Joong Kim and Jong-Hee Chae are the editorial board members of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: WJK, SYK, AC, and JHC. Data curation: HW, SL, and JYH. Formal analysis: HW. Methodology: WJK, MJK, MWS, and JHC. Project administration: HW and JHC. Visualization: HW, SYK, and AC. Writing-original draft: HW. Writing-review & editing: BCL, KJK, and JHC.