Introduction
Wieacker-Wolff syndrome (WWS) (WRWF [OMIM #314580]; female restrictive type known as WRWFFR [OMIM #301041]) was first reported as being characterized by congenital contracture, slowly progressive distal muscle atrophy, dyspraxia of the eye, face, and tongue muscles, and mild intellectual impairment. This disease was initially described by monitoring disease progression in six men from three generations in one family, and an X-linked recessive genetic pattern was observed in 1985 [1]. Later, Kloos et al. [2] revealed that the causative gene of WWS was located at Xq11.2. In 2013, Hirata et al. [3] investigated the causative gene of WWS, discovering the zinc-finger gene zinc finger C4H2-type containing (ZC4H2) as pathogenic using next-generation sequencing (NGS) and array comparative genomic hybridization in patients with X-linked intellectual disability (ID) and arthrogryposis multiplex congenita (AMC). In 1991, exotropia, microcephaly, distal muscle wasting, and digital arch were observed in one family, which was reported at the time as having Miles-Carpenter syndrome [4]. In 2015, May et al. [5] revealed this to be a case of ZC4H2 related-disease via genetic evaluation. This familial disease was thereafter classified as the same disease and referred to as “ZC4H2-associated rare disease” (ZARD) [6].
The ZC4H2 protein comprises a zinc finger domain, four cysteine residues, two histidine residues, and a coiled-coil region. The coding gene, ZC4H2 is located on the long arm of the X chromosome (Xq11.2). To date, various phenotypes of WWS have been reported, with symptoms including facial dysmorphism, skeletomuscular symptoms involving multiple joint contractures, and neurologic symptoms including ID and motor delay [4-13]. In addition, several recent studies have reported that the female phenotype can be particularly severe [6-10].
Here, we present a summary of various clinical phenotypes associated with ZC4H2 in six patients.
Materials and Methods
In this study, six patients diagnosed with diseases related to a ZC4H2 gene variant who visited a pediatric neurologic outpatient clinic at the Seoul National University Hospital Children’s Hospital are reported.
For clinical phenotyping, a retrospective chart review was conducted, as well as an analysis of brain magnetic resonance imaging (MRI), and electroencephalography (EEG) findings. Based on NGS analysis, SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA), a capture probe targeting the entire exonic region was used for exome sequencing. Library preparation was performed in accordance with the manufacturer's instructions. Libraries were sequenced (paired-end) using the HiSeq 2500 sequencing system (Illumina, San Diego, CA, USA). For sequencing, paired-end sequence reads with a read length of 151 base pairs were aligned to the Genome Reference Consortium Human Build 37 (GRCh37) using the Burrows-Wheeler Aligner (v. 0.7.17). Picard software (v. 2.9.0), SAMtools (v. 1.9), and Genome Analysis Toolkit (v. 4.1.2) were used for deduplication, realignment, and basic recalibration. We used a different haplotype, Caller, to perform the calling. SnpEff, ANNOVAR, and InterVar were used to annotate transformations. In the absence of a specific trait gene, a variant with an alternate allele frequency (AF) of 0 in the gnomAD database was considered a candidate autosomal dominant gene. AF variants of less than 0.01% were considered candidates for autosomal recessive genes. For low-frequency strain detection, we used MuTect2 to search for variants with an AF between 0.05 and 0.25. Only low-frequency variants with an allele depth of 30 or more and 10 or more variant alleles were selected. For copy number variation (CNV) analysis, chromosomal microarray (CMA) assays were performed using the Agilent Human Genome Oligonucleotide Comparative Genome Hybridization 244, 80, or 60 K microarrays (Agilent Technologies), with median total probe spacings of 8.9,13, or 41 kb, respectively. All CNVs were called based on human GRCh37 (hg19).
Quantitative polymerase chain reaction (qPCR) was performed to differentiate germline mosaics using ribonuclease P (RNase P) as the reference gene. All variants were categorized according to the standards and guidelines of the American College of Medical Genetics and Genomics, and pathogenic or likely pathogenic variants were defined as causative variants.
This study was approved by the Institutional Review Board (IRB) of Seoul National University Hospital (IRB No. 1406-081-588) for whole-exome sequencing and a review of medical records. All patients or their legal representatives provided written informed consent.
Results
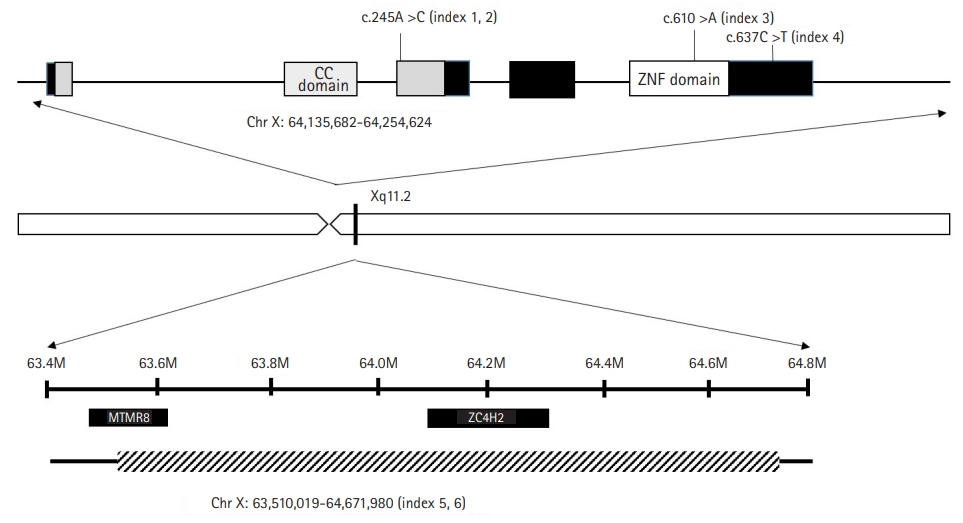
Four of the enrolled patients were male and two were female. Four patients were diagnosed using NGS through the Korean Undiagnosed Disease Program, and three missense variants were identified (Table 1). None of the variants confirmed by Sanger sequencing were found in the 1,000 Genomes Project or the gnomAD database. All missense mutations, c.245A>C (p. Gln82Pro), c.610C>A (p. Pro204Thr), and c.637C>T (p. Arg213Trp), were inherited from the mother. c.637C>T has been reported previously, and the other two variants are novel [3]. The female patients were dizygotic twins, in whom we identified a 1.16-Mb deletion including the ZC4H2 gene in Xp11.2 by CMA.
These patients had a wide variety of clinical manifestations (Table 2), with central or peripheral neurological symptoms being the most prominent phenotype. The clinical features were analyzed with a focus on previously reported phenotypes, according to research stating that 30% of ZARD cases were seen in 42 families (Frints et al. [6]). All patients experienced motor delays. Other findings included ID (6/6), poor speech (6/6), inability to walk (4/6), spasticity (3/6), seizures (3/6), and hyperreflexia (1/6). Brain MRI was performed in five patients, except for index case 6, and only index case 4 showed myelination delay, with no major abnormalities. EEG was performed on three patients who had seizures. In index case 1, generalized spikes and waves were observed, and seizures were controlled by taking two anticonvulsants (valproic acid and levetiracetam) due to frequent recurrence. In index case 2, convulsions occurred only once around 1 year of age and were observed normally on EEG at the time; the patient was observed without taking anticonvulsants. Index case 4 showed intermittent generalized slow activity without definite epileptiform discharges on EEG, but there was no seizure after the first seizure.
Craniofacial symptoms included microcephaly (2/6), low-set ears (3/6), a U-shaped upper lip vermilion (3/6), short neck (2/6), and microretrognathia (2/6). Some patients also exhibited ocular symptoms, such as strabismus (3/6), ptosis (2/6), and ocular motor apraxia (2/6). Five of the six patients had musculoskeletal phenotypes. The symptoms included multiple arthrogryposis: metacarpophalangeal joint contracture (4/6), clubfoot (3/6), distal muscle weakness (3/6), other Achilles tendon contractures (3/6), knee flexion contractures (2/6), camptodactyly (2/6), elbow flexion contractures (1/6), and hip subluxation (2/6). Other clinical symptoms included short stature (2/6), recurrent aspiration pneumonia (3/6), and feeding difficulty during infancy (3/6).
Discussion
The zinc-finger gene ZC4H2 was first identified by Hirata et al. [3] in 2013, and it was confirmed to be strongly expressed in the brain and spinal cord of both mice and zebrafish. In mice, Zc4h2 was revealed to play an important role in brain development, being strongly expressed in the embryo stage, with a decrease in the postnatal stage; knockdown of zc4h2 in zebrafish results in impaired swimming and a reduced number and disorganized pattern of neuromuscular endplates. Based on these findings, it was concluded that pathologic variants of the ZC4H2 gene cause AMC and ID due to impairment of central and peripheral synaptic plasticity. In 2015, May et al. [5] reported that homozygous zc4h2 mutants resulted in an abnormal swing capacity, pectoral fin flexion, and eye position in zebrafish.
Although the first known and characteristic clinical features of WWF are AMC and ID, this condition is now referred to as ZARD because of its various clinical expressions. The patients in our cohort often showed one of the features of AMC and ID, or a clinical variation other than those two symptoms. Two female patients showed a global developmental delay of 24 months, but all other patients were diagnosed with ID, and three patients had AMC. In addition, relatively unknown facial dysmorphism, ocular symptoms, and gastrointestinal and respiratory symptoms such as poor feeding and aspiration pneumonia were also observed.
All four male patients had missense mutations that were transmitted from asymptomatic mothers. Index cases 1 and 2 showed the same mutation, c.245A>C (p.Gln82Pro), and the clinical symptoms also showed similar facial dysmorphisms, including a high forehead, low-set ears, ocular motor apraxia, and a long philtrum in both patients. Musculoskeletal symptoms were not clear, and other neurologic phenotypes, such as inability to walk, motor delay and ID, poor speech, and seizures, were observed. The missense variant c.187G>C (p.Val63Leu), found in the coiled-coil domain, was identified in a family containing six patients, all of whom showed ptosis, contracture of the Achilles tendon, distal muscle weakness, motor delay, and ID [3]. In both these mutations, AMC, the most well-known characteristic of ZARD, was relatively inconspicuous or showed only relatively mild musculoskeletal symptoms such as metacarpophalangeal joint contractures. Although a previous study revealed that a point mutation in the coiled-coil domain can cause destabilization of the ZC4H2 protein, there has been no study on the exact function of the coiled-coil domain to date [5].
Index case 3 had a c.610C>A (p.Pro204Thr) mutation, which is located in the zinc figure domain (Fig. 1). Unlike the previous two patients, the musculoskeletal symptoms of AMC, clubfoot, Achilles tendon contracture, and distal muscle weakness were prominent. Many other studies have reported missense mutations in this area and associated characteristic musculoskeletal symptoms and ID, including AMC [3,11].
Index 4 had the c.637C>T (p.Arg213Trp) variant, which is located in the C-terminus, and has been identified as a pathologic variant in previous reports [3]. Similar to a previously reported patient with the same mutation, this patient also did not have musculoskeletal symptoms, and central or peripheral neurologic symptoms such as motor delay, ID, seizure, and spasticity were more definite.
Although the exact function of each domain has not yet been determined, considering our patients and other previously reported patients, missense mutations in the zinc finger domain appear to be prominently associated with musculoskeletal symptoms such as AMC.
However, all previous studies, including large-scale studies that investigated several families, showed several clinical variability even with the same genotype, so the genotype-phenotype (G-P) correlation could not be clearly identified [3,6,11]. However, in our cohort, the phenotypes were different for each protein region. If further studies identify more patients with ZARD, that would be of great help in revealing common G-P correlations.
Previous reports have shown that female carriers can be asymptomatic or almost unaffected, but a missense mutation, frameshift mutation, splicing variant, and Xq11.2 microdeletion caused by de novo changes had mild to severe impacts when ZC4H2 was completely or partially lost [6-10,13]. In a study by Frints et al. [6], which analyzed the difference between de novo mutations and those transmitted by carrier females in 24 families, 12 of the carrier females (n=33) showed a normal phenotype, and the remaining 20 had mild ID or joint contractures (phenotype limited to fingers, wrists, etc.). In contrast, more than 90% of the patients with de novo pathologic phenotypes showed central and peripheral neurologic symptoms, such as clinical presentation, AMC, motor delay, inability to walk, distal muscle weakness, and more characteristics of ZARD
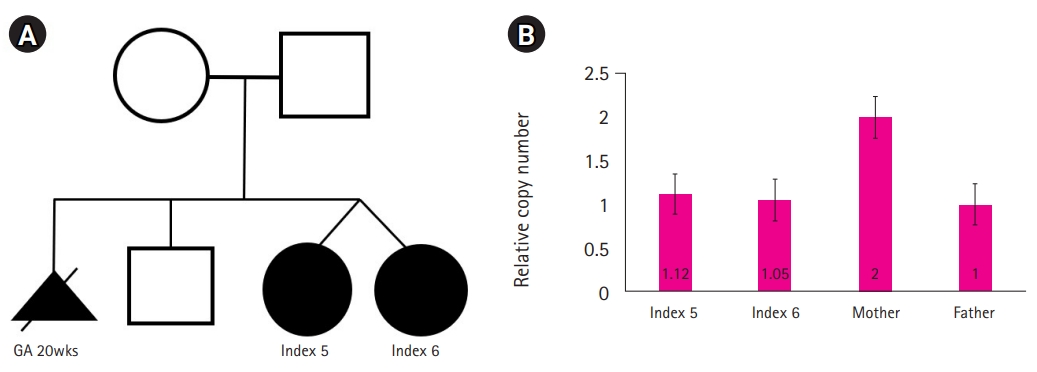
Our cohort included two female patients. These patients were dizygotic twins harboring the same deletion (X: 63510019-64671980). In the twin female patients, the entirety of the ZC4H2 gene and part of the myotubularin related protein 8 (MTMR8) gene were included in the deletion. The MTMR8 gene is known to be related to X-linked myotubular myopathy, making it difficult to describe the patient's phenotype [14].
Interestingly, in these two patients, other benign CNVs were found in different forms. Parental tests were performed using qPCR, a target-specific detection method, which found that neither the father nor mother harbored this deletion (Fig. 2).
The mother of the dizygotic twins had previously selectively aborted her first fetus with fetal hydrops at a gestational age of 20 weeks, and had subsequently given birth to a healthy boy. The known prenatal clinical features of ZARD include clubfoot, hypokinesia/akinesia, rocker-bottom foot contractures, AMC, and edema [6,13]. Since the aborted fetus was not tested for Xp11.2 microdeletion, it is not known whether fetal hydrops was a prenatal manifestation. The second and third children, index cases 5 and 6, had decreased fetal movement, showed facial dysmorphisms (e.g., a small mouth and retracted chin), and AMC, and suffered respiratory difficulties due to a short neck and laryngomalacia. Although both patients carried the same deletion, index case 5 showed a more severe phenotype than index case 6, which is consistent with other studies showing variable phenotypes arising from the same variant [3,6].
In the paternal test, the parents were normal, but the deletion of the same region in both fraternal twins was observed. Furthermore, the details of the previously aborted fetus gave rise to suspicion of a history of ZARD. Although invasive tests, such as ovarian biopsy, were not performed to confirm this speculation, germline mosaicism rather than a de novo mutation was considered. In the absence of such reports previously, it is not possible to compare the severity of de novo mutations with expression due to germline mosaicism. Nonetheless, more cases and additional laboratory studies on these germline mutations are required.
Although not implemented in our study, other studies have attempted to prove the severe phenotype in de novo females via the X-inactivation test [6-8]. However, since several studies have shown inconsistent results, this cannot be clearly determined as a phenomenon caused by X-inactivation.
In conclusion, ZARD is well known to present with severe AMC and ID, mainly in males, but none of the patients showed the same phenotype. Phenotypic severity can vary. Since ZARD is an X-linked recessive disease, most female patients with conserved carriers are asymptomatic or have mild symptoms; however, female patients with deleterious mutations showing severe symptoms were also observed. Further research is needed to clarify the phenotypic variability and the molecular and cellular mechanisms of this rare disorder.