Epilepsy with SLC35A2 Brain Somatic Mutations in Mild Malformation of Cortical Development with Oligodendroglial Hyperplasia in Epilepsy (MOGHE)
Article information

Abstract
Purpose
This study presents the characteristics of patients with mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE) with SLC35A2 somatic variants in the brain who underwent epilepsy surgery and showed clinical improvement in seizures.
Methods
We collected 10 patients with SLC35A2 somatic mutations in the brain who underwent surgery to treat drug-resistant epilepsy at Severance Children’s Hospital from 2014 to 2019 and retrospectively reviewed their genetic profiles, neuropathologic results, clinical features, pre-operative evaluations, and post-operative outcomes.
Results
Six of the 10 patients with SCL35A2 somatic mutations in the brain had Lennox Gastaut syndrome (LGS) evolving from infantile spasms (IS), three had LGS, and one had IS. The median value of variant allele frequencies (VAFs) was 5.7% (1.7% to 5.8%; range, 1.4% to 22.9%). Nonsense mutations were the most common (50%), followed by missense mutations (40%) and a splicing site mutation (10%). Eight patients (80%) had good post-operative outcomes, with freedom from disabling seizures in five (Engel class I) and rare disabling seizures in three (Engel class II). Four of the eight patients who could be assessed for social quotient (SQ) after surgery showed SQ improvements by 12.2±6.4. Although all patients were finally diagnosed with MOGHE, seven (70%) were initially diagnosed with gliosis, two with mild malformation of cortical development, and one with no abnormality.
Conclusion
All patients with SCL35A2 brain somatic mutations, even with low VAFs, had refractory epilepsy such as LGS or IS, and were finally diagnosed with MOGHE. This report is the first in Korea to our knowledge.
Introduction
Malformations of cortical development (MCDs) refer to a wide range of cortical lesions from disruption of neurogenesis, proliferation, apoptosis, or migration [1,2]. The concept of MCDs was introduced in pediatric patients with developmental delay and epilepsy. A classification of those malformations was proposed in 1996 [3]. This classification was revised and updated in 2012 including mild MCD (mMCD) or focal cortical dysplasia (FCD), which features a small proportion of abnormal brain cells and disorganized cortical lamination [4]. Furthermore, mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE), which is characterized by increased proliferation of oligodendroglia in the white matter and deep gray matter, was proposed as a new histopathologic entity in 2017 [5].
Ample evidence has established that FCD is associated with intractable epilepsy requiring surgical resection of the lesion [1,6]. Remarkable advances in genetic technologies and molecular biology have revealed that somatic mutations in brain cells are associated with FCD [7,8]. Moreover, a low-level brain somatic mutation burden—even with a variant allele frequency (VAF) less than 1%— is enough to cause intractable epilepsy [9,10]. Therefore, deep sequencing with a high read depth (more than 1,000×) is necessary to identify the causative gene variant [10,11]. Genes related to epilepsy have been identified, including SLC35A2 and genes encoding the mechanistic target of rapamycin (mTOR) pathway proteins (AKT3, DEPDC5, MTOR, PIK3CA, TSC1, and TSC2) [8,10,11].
SLC35A2, which is located at Xp11.23, encodes a uridine diphosphate (UDP)-galactose transporter, a member of the nucleotide-sugar transporter family that transports galactose from the cytosol or nucleus into Golgi vesicles [9,12]. Germline loss of function variants in SLC35A2 resulting in producing abnormal truncated glycans that lack galactose have been identified in multiple patients with epileptic encephalopathy [13,14]. In addition, brain mosaic mutations in SLC35A2 are now considered one of the major genetic causes of intractable epilepsy, and recent studies have reported the histopathologic features of SLC35A2 somatic variants [12].
In this study, we present the clinical characteristics of patients with SLC35A2 somatic mutations in the brain who were finally diagnosed with MOGHE.
Materials and Methods
1. Selection of subjects
We retrospectively collected a series of patients with intractable pediatric epilepsy who underwent epilepsy resection surgery and were confirmed to have SLC35A2 brain somatic mutations from January 2013 to December 2019 at the Epilepsy Research Institute of Severance Children’s Hospital. Ten cases were enrolled, including six patients (patient numbers 1, 2, 3, 4, 5, and 7) who were previously reported in 2018, three patients (patient numbers 8, 9, and 10) reported in 2019, and one patient (patient number 6) reported in 2021 [10,12,15]. We reviewed all data available for these 10 patients, which comprised their demographic characteristics, clinical features, pre-operative investigations, genetic profiles, operation details (including the pathologic analysis), and post-operative outcomes with medical records.
All patients were informed about the study and agreed to the collection of human tissues, and the protocols were approved by Severance Hospital and the KAIST Institutional Review Board and Committee on Human Research (IRB No. 4-2017-0119).
2. Pre-operative evaluations
The pre-operative investigations included neurologic examinations, development tests, and evaluations for determining the surgical area. The epileptogenic area was determined through the interpretation of long-term video-electroencephalogram (EEG)-monitoring data and confirmed by imaging studies including high-resolution magnetic resonance imaging (MRI), fluoro-deoxy-glucose (FDG) positron emission tomography (PET), and subtraction ictal single-photon emission computed tomography (SPECT) co-registered to MRI (SISCOM).
The development level was measured considering both cognitive level and social function, and it was classified as follows: normal (intelligence quotient/developmental quotient >70), mild delay (50 to 70), moderate delay (35 to 49), severe delay (20 to 34), and profound delay (<20). The cognitive level was assessed using the Korean Bayley Scales of Infant Development-II, the Korean Wechsler Preschool & Primary Scale of Intelligence-IV or the Korean Wechsler Intelligence Scale for Children-IV according to the patient’s age. The social quotient (SQ), as an indicator of general adaptive function, was scored in all patients using the Korean version of the Social Maturity Scale (SMS) based on the Vineland Social Maturity Scale, fifth version.
3. Genetic profiles and pathologic analysis through brain samples
Surgery proceeded in two stages: inserting intracranial EEG and determining the surgical margin according to the protocol of the Epilepsy Research Institute, Severance Children’s Hospital [10,16]. All brain samples were freshly frozen (FFZ) or formalin-fixed paraffin-embedded (FFPE).
For DNA extraction from brain tissue, we used QIAamp DNA Mini kits (Qiagen, Germantown, MD, USA) from FFZ brain samples and QIAamp DNA FFPE kits (Qiagen, Hilden, Germany) from FFPE, as described in the previous report [10]. Site-specific amplicon sequencing for genetic analysis of SLC35A2 with read depth >100,000× and region-specific primers with the Illumina Nextera single index (Illumina, San Diego, CA, USA) for validation sequencing for somatic mutations was performed, as previously described [10,15].
Histopathologic analyses were conducted twice; the first one was done at the time of surgery by the Department of Pathology at Severance Children’s Hospital with the International League against Epilepsy (ILAE) classification [17]. Because MOGHE was first introduced in 2017 [5] and has not yet been included in the ILAE classification, a reanalysis was performed with hematoxylin and eosin staining, as well as anti-NeuN to look for heterotopic neurons (Clone A60, Millipore, Temecula, CA, USA) and anti-olig2 to identify oligodendroglial cells (clone JP18953, IBL International, Hamburg, Germany) immunostaining. The reanalysis was conducted by an expert neuropathologist without knowing the genetic information and previous pathologic results at Schoen Klinik, Vogtareuth, Germany in 2020 [12].
Results
We enrolled a total of 10 patients with intractable pediatric epilepsy confirmed to have SLC35A2 brain somatic variants.
1. Demographics and clinical features
As shown in Table 1, six of the 10 patients were male (60%). All patients suffered from daily seizures and had refractory epilepsy with focal epileptic findings on EEG, making them candidates for epilepsy surgery. The mean age of seizure onset was 12.2±12.1 months old (range from 3 months to 3 years 2 months of age). Seventy percent of patients were diagnosed with infantile spasms presenting with spasms, and six of those patients progressed to Lennox Gastaut syndrome (LGS) with various types of seizures, including focal impaired-awareness seizures, focal motor seizures, and myoclonic seizures. Thirty percent of cases had only LGS, of whom two (2/3) experienced generalized seizures with head drop and one (1/3) had focal impaired-awareness seizures and spasms.
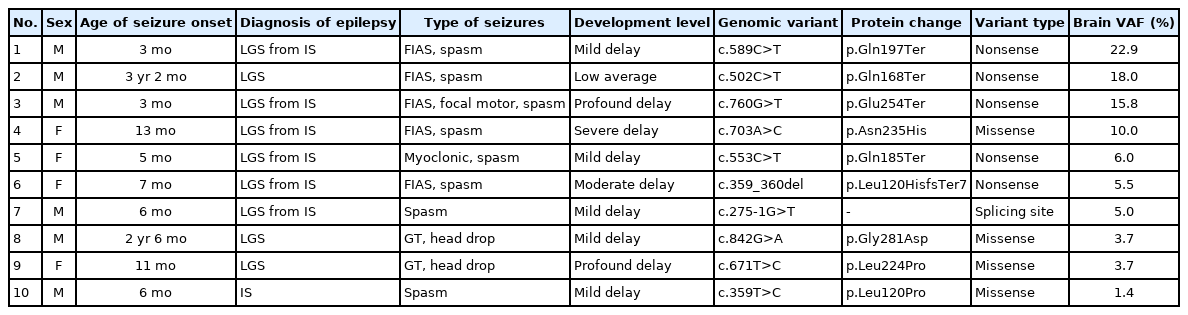
Demographics, clinical features, and genetic profiles
The degree of developmental delay was determined through an analysis of the cognitive level and social function; 10% of patients had low average development, 50% showed mild delay, 10% had moderate delay, 10% presented severe delay, and 20% had profound delay. The average SQ was 45.7±22.2, corresponding to a moderate delay.
2. Genetic profiles
SLC35A2 brain somatic mutations were confirmed by site-specific amplicon sequencing (read depth >100,000×). Nonsense mutations were found in 50% of patients, missense mutations in 40%, and a splicing site mutation of 10%. The SLC35A2 VAFs ranged from 1.4% to 22.9% (median VAF, 5.7% [range, 1.7% to 5.8%]), and patients were assigned numbers based on the VAFs in descending order. Patients 1, 2, and 3, who had high VAFs (more than 15%), all had nonsense mutations, while patients 8, 9, and 10, who had low VAFs (less than 5%), were all identified as having missense mutations (Table 1).
3. Pre-operative evaluations, operation details, and post-operative outcomes
As presented in Table 2, four patients (40%) underwent epilepsy surgery twice, three of whom (3/4) had resection surgery after localization of the lesion following corpus callosotomy. All these patients initially had negative findings on MRI, but localization by EEG was possible after the first operation. The mean age at the final operation was 5.4 years (range, 2 years and 1 month–10 years and 2 months of age).

Pre-operative evaluations, operation details, and post-operative outcome
Long-term video-EEG monitoring findings by both interictal and ictal localization demonstrated multiple ictal onset zones in three patients (30%). Three cases (30%) had cortical abnormality findings from pre-surgical MRI. All of them had previous MRI findings characteristic of MOGHE [18]; patient 5 had FCD on the right frontal lobe, patient 7 had a slightly increased white matter T2 signal around the right temporo-parietal lobe, and patient 8 had FCD on the left frontal lobe. FDG-PET was performed in all patients, and the hypometabolic lesions were concordant with the EEG results in seven patients (70%), showing more extensive lesions. SPECT was performed in only five patients because the other patients had an insufficient seizure duration to carry out the tests. SISCOM was obtained in only two of these patients, and all results were consistent with EEG localization.
The initial histopathologic diagnosis at the time of surgery was gliosis in 70%, mMCD in 20%, and normal findings in 10%. However, after a pathologic reanalysis in Germany in 2020, all cases were confirmed as MOGHE, featuring increased olig-2 immunostained cells not only in the gray-white matter junction, but also in the deep white matter, and heterotopic neurons in the white matter as previously described [5,12].
The mean follow-up period was 3.4 years (range, 11 months to 6 years and 3 months), and patients took an average of 1.8 anti-seizure medications (range, 0 to 4 at the time of the last follow-up). Eighty percent of cases reached Engel class I–II with complete freedom from seizures or rare disabling seizures, while the 20% of cases with the lowest VAFs (patients 9 and 10) showed worthwhile seizure reduction of Engel class III according to the Engel Epilepsy Surgery Outcome Scale [19]. Post-operative developmental tests were performed for eight patients, but as their age changed after surgery, it was difficult to make simple comparisons using a different tool from the test performed in a pre-operative cognitive evaluation. Nonetheless, the SMS was administered to all patients before and after the operation, and four patients showed improvement by an average of 12.2±6.4 points in the SQ level.
Discussion
In this study, we presented the clinical features of patients with SLC35A2 brain somatic variants showing the pathologic characteristics of MOGHE. SCL35A2 somatic variants have been reported in correlation with neuropathologic phenotypes with MOGHE [12]. The definition, classification, clinical phenotypes, MRI findings, pathology, and correlated genetic variants of MCD remain a challenging topic, and updates in this field continue [20]. As the concept of MOGHE has been introduced relatively recently, its clinical importance is underestimated in practice [5].
In our study, most of the mutations in SLC35A2 were nonsense mutations (50%), including one frameshift deletion, and this category included the three patients with the highest VAFs. Brain tissues with the p.Gln197Ter variant from patient 1 and the p.Glu254Ter variant from patient 3 showed aberrant patterns in terms of pathogenicity, and patient 2, with the p.Gln168Ter variant, showed changes in UDP-galactose transport as previously reported [13,15]. SLC35A2 is involved in transporting UDP-galactose from the cytosol to the Golgi apparatus and completing glycosylation by attaching galactose to N-acetylglucosamine. Loss of function of SLC35A2 can cause problems with N-glycosylation in this process. Abnormal N-glycosylation of brain development affects neural transmission, myelination, and neuronal migration, leading to clinical neurologic symptoms such as congenital disorders of glycosylation (CDG) [21], and the phenotype of MOGHE involves increased heterotopic neurons in the white matter and oligo-2 positive cell clusters in the white matter and gray-white matter junction. Ultimately, these findings appear as a blurred gray-white matter junction and hypomyelination on brain MRI. Most patients in our study presented normal MRI findings (70%), while three patients had increased T2 signal intensity on the lesion. The MRI findings of MOGHE are known to involve an increased laminar signal at the corticomedullary junction on T2 and fluid attenuated inversion recovery in subtype I in younger children or an increased signal of the adjacent white matter in subtype II in older children [18]. Therefore, somatic brain variants of SLC35A2, which likely occur in a neuroglial progenitor cell during neurogenesis, are thought to have potential as a genetic marker for MOGHE [12].
In this study, the three patients with the low VAFs (less than 5%) were all missense mutations, which means that a low mutation burden is sufficient to cause intractable epilepsy. Of particular note, patient 10 with a VAF of 1.4% and patient 9 with a VAF of 3.7% belonged to Engel class III, with only partially controlled seizures after epilepsy surgery. The variant burden in the brain may also govern aspects of the clinical presentation, but this mechanism is still unclear [9]. Further research is needed to determine whether the VAF in a fraction of cells in the blood is proportional to the VAF in brain somatic mutations and could be used as a factor to determine clinical severity.
Considering the reports that the symptoms of SLC35A2-CDG patients improved after taking galactose supplementation, which may partially increase cytosolic UDP-galactose and thereby facilitate galactosylation through alternative UDP-galactose transport into the Golgi [12,22], precision treatment can be considered to control the symptoms of patients with SLC35A2 somatic brain mutations who cannot undergo epilepsy surgery or continue to have seizures after surgery.
Notes
Hoon-Chul Kang is an associate editor, Ara Ko, Se Hee Kim, Joon Soo Lee and Heung Dong Kim are the editorial board members of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: SHK, JSL, HDK, and HCK. Data curation: HJK, DSK, SHK, JHL, AK, and HCK. Formal analysis: HJK, AK, SHK, JSL, and HCK. Funding acquisition: HDK and HCK. Methodology: DSK, SHK, JHL, AK, SHK, JSL, HDK, and HCK. Project administration: HJK. Visualization: HJK. Writing-original draft: HJK. Writing-review & editing: HDK and HCK.
Acknowledgements
This study was supported by the Team Science Award of Yonsei University College of Medicine (6-2021-0007) and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI21C1659).