X-Linked Cerebral Creatine Deficiency Syndrome with Prolonged QT Interval: A Case Report
Article information

Creatine is a metabolite that plays an important role in maintaining brain, heart, and muscle function [1]. It is synthesized in the kidney, liver, and pancreas by arginine glycine acyltransferase (AGAT, chromosomal location 15q15.1) and guanidinoacetic acid methyl transferase (GAMT, chromosomal location 19p13.3), and it is transported to the brain and muscle by the creatine transporter SLC6A8 [1]. Creatine is metabolized by creatine kinase to produce adenosine triphosphate, which maintains organ function [1]. Cerebral creatine deficiency syndromes (CCDS) are classified into three types: two autosomal recessive types, in which mutations in the peptide sequence of either AGAT or GAMT disrupt creatine synthesis, and an X-linked type, in which the creatine transporter SLC6A8 is deficient [2]. Intellectual disability, seizures, and speech delays are commonly observed in CCDS-affected individuals [3]. Intractable epilepsy and early global developmental delay with autistic behavior have also been observed in severe cases [3]. The diagnosis of CCDS is based on the measurement of guanidinoacetate, creatine, and creatinine in plasma and urine, as well as genetic testing [1]. Creatine deficiency in the brain detected by proton magnetic resonance spectroscopy (1H-MRS) is a characteristic finding of CCDS [1]. Here, we describe a case of CCDS caused by creatine transporter deficiency presenting with long QT syndrome and a large arachnoid cyst in the frontal lobe.
A 7-year-old boy presenting with bilateral tonic-clonic seizures, intellectual disability, and hyperactivity was referred to our pediatric clinic. He had been delivered at 40 weeks of gestation without pre- or post-partum complications. The birth weight was small for the gestational age (2,600 g, third percentile). The parents were healthy and nonconsanguineous, with no history of neurological or genetic disorders. The patient had a history of complex febrile seizures starting at 14 months of age with cognitive, speech, and motor delays. He could not speak any words until the age of 7 years. A physical examination revealed a short stature (114.6 cm, second percentile), low body weight (15.8 kg, less than the first percentile), and microcephaly (48 cm, less than the first percentile). The patient had dysmorphic facial features, including a triangular face, flat forehead, downward slanting palpebral fissures, and protruding mouth. Laboratory findings (complete blood count, serum electrolyte and glucose concentrations, hepatic and renal function tests, and routine urinalysis) were normal. Metabolic tests for lactic acid, creatine kinase, ammonia, plasma amino acids, and urine organic acids were normal; but a test for thyroid function revealed hypothyroidism. A large arachnoid cyst in the right frontal lobe was identified via brain magnetic resonance imaging (MRI) (Fig. 1A); however, electroencephalography (EEG) results were normal. Peripheral blood chromosome analysis and a chromosomal microarray test revealed no abnormalities. Whole-exome sequencing (WES) was performed to identify potential genetic abnormalities that could cause intellectual disability, seizures, and dysmorphic facial features. Written informed consent was obtained from the parents, and the study was approved by the Institutional Review Board of Busan Paik Hospital (Busan, Korea; approval number: 2021-07-002). The results of WES revealed the presence of a hemizygous frameshift SLC6A8 variant, NM_005629: c.626_627delCT (p.Pro209ArgFs*87) that was also found to be carried by the patient’s mother. This result was confirmed by direct sequencing (Supplementary Fig. 1). Since there was no specific family history in the maternal family and no family members showed phenotypes such as intellectual disability or seizures, additional genetic testing of the maternal family was not performed. The patient’s brain 1H-MRS revealed a decreased creatine peak in the corona radiata and basal ganglia (Fig. 1B). He was therefore diagnosed with an X-linked creatine transport disorder caused by a pathogenic variant of SLC6A8 and was started on drug therapy comprising creatine monohydrate (100 mg/kg/day), L-arginine (400 mg/kg/day), and L-glycine (150 mg/kg/day). These supplements failed to improve his cognitive or behavioral problems and were eventually discontinued.
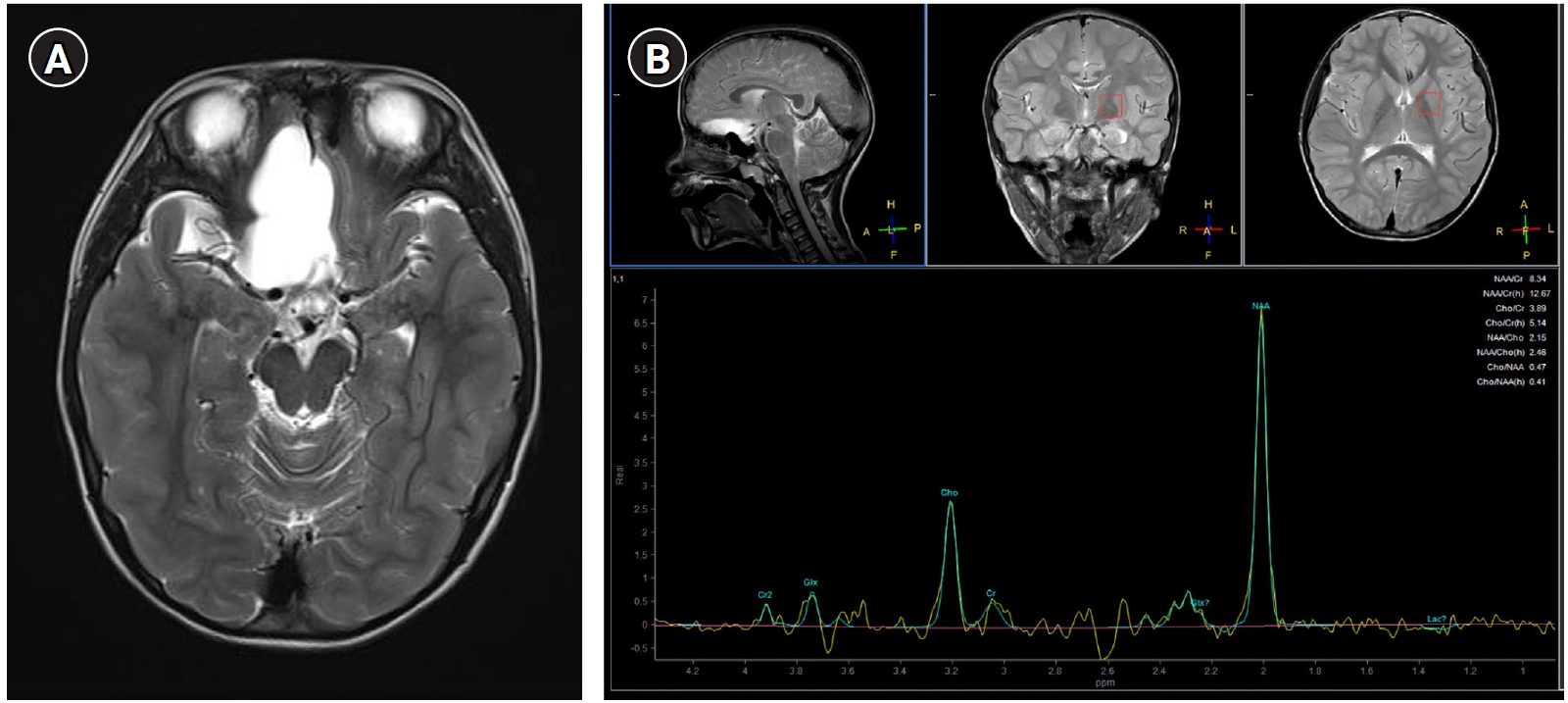
(A) T2-weighted axial brain magnetic resonance imaging showing a focally enlarged arachnoid cyst located in the right inferomedial frontal area. (B) Proton magnetic resonance spectroscopy results showing a decreased creatine peak in the regions of interest of the corona radiata and basal ganglia.
At age 9, the patient experienced repetitive bilateral tonic-clonic seizures that were well controlled with oxcarbazepine treatment. A new brain MRI revealed that the arachnoid cyst had increased in size and displaced the right optic nerve, but an ophthalmologic examination was normal, and an additional brain imaging session was scheduled for a year in advance as follow-up, without any neurosurgical treatment. EEG results identified intermittent spikes in the right frontal area. Unexpectedly, a prolonged QT interval (QTc time 488 ms; reference range, 350 to 440) was detected on an electrocardiogram (ECG) (Fig. 2). The patient had never been treated with any QT-prolonging drug. Long QT syndrome was finally diagnosed following a test with a Holter monitor. The patient was placed under close observation, but no beta-blocker medication was prescribed due to his young age.

Electrocardiogram results showing a prolonged QT interval (QTc 488 ms).
The CCDS diagnosis in this patient presented difficulties because the clinical manifestations, such as seizures, intellectual disability, and facial dysmorphism, were non-specific. As we were unable to find an etiology for our patient using conventional cytogenic methods, chromosomal microarray, or metabolic tests, we resorted to WES and were able to make a diagnosis of creatine transporter deficiency caused by a frameshift variant in SLC6A8. His mother was a carrier of this variant. After genetic diagnosis, brain 1H-MRS demonstrated a specific decrease in creatine levels in the brain of the patient. Analyses of urinary guanidinoacetate and the creatine/creatinine ratio are important screening tests for all CCDSs [1]. In patients with SLC6A8 deficiency, the increase in urinary creatine excretion together with the inherently low urinary creatinine excretion results in an elevation of the urinary creatine/creatinine ratio, which serves as a valuable diagnostic marker in male patients [4,5]. We could not measure urinary creatine levels in our patient because this test was not available at our institution. However, the decreased creatine level in the brain and genetic testing revealing a mutation in SLC6A8 were sufficient to confirm the diagnosis of creatine transporter deficiency.
Long QT syndrome was also detected from the prolongation of the QTc interval in our patient’s ECG results. Several genes (KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2, among others) are associated with long QT syndrome [6]. However, none of these genetic mutations were detected in our patient. A few studies have reported cardiac syndromes in patients with creatine transporter deficiency. van de Kamp et al. [7] described one patient with long QT syndrome, two patients with mild cardiomyopathy, and one patient with multiple premature ventricular contractions in their retrospective study of 101 male individuals with X-linked creatine transporter deficiency. Levin et al. [8] recently reported seven patients with QTc prolongation in a sample of 18 patients with creatine transporter deficiency. The same authors also demonstrated that long QTc was associated with cardiac dysfunction and sudden death in an animal model for creatine transporter deficiency (Slc6a8−/y mice). Because expression of the SLC6A8 gene is high in skeletal muscle, heart, and kidney, cardiac problems could be expected in patients with creatine transporter deficiency [9]. Although the exact mechanism is currently unknown, it can be hypothesized that the SLC6A8 gene is somehow involved in the physiology of heart muscle. Therefore, cardiac evaluations such as ECG and echocardiography are necessary in children with creatine transporter deficiency to detect potential cardiac involvement.
CCDS treatment can correct cerebral creatine depletion [2]. Oral supplementation of creatine can be administered to maximize the delivery of creatine into the brain, together with creatine precursors such as L-arginine and L-glycine [2]. However, the value of creatine supplementation in patients with creatine transporter deficiency is unclear because delivery into the brain is not efficient enough to fully compensate for the creatine depletion [2]. Our patient failed to show any clinical improvement in behavior, attention, or cognitive problems associated with the syndrome in response to creatine, L-arginine, and L-glycine supplementation. Since there may be some residual transporter activity, creatine supplementation is generally recommended for all new patients, but definite success has not been reported [1].
To our knowledge, this is the first case of creatine transporter deficiency reported in Korea. Based on our experience, we conclude that the urinary creatine/creatinine ratio and brain MRI scanning with spectroscopy can be useful tools to screen male patients with intellectual disability and seizures of unknown origin for creatine transporter deficiency. Furthermore, it is to be expected that advances in next-generation sequencing will facilitate the diagnosis of creatine transporter deficiency. Cardiac evaluation and monitoring are necessary because these patients are likely to have cardiac problems such as long QT syndrome and cardiomyopathy.
Supplementary materials
Supplementary materials related to this article can be found online at https://doi.org/10.26815/acn.2022.00031.
Sanger sequencing electropherograms revealing a hemizygous frameshift variant (c.626_627delCT) of the SLC6A8 gene carried by the patient’s mother.
Notes
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: KSL and BLL. Data curation: HWK and BLL. Methodology: JKP, JEL, KSL, and BLL. Project administration: HWK and BLL. Visualization: HWK and BLL. Writing-original draft: HWK and BLL. Writing-reviewing & editing: HWK, JKP, JEL, KSL, and BLL.