An 8q24.11q24.13 Microdeletion Encompassing EXT1 in a Boy with Autistic Spectrum Disorder, Intellectual Disability, and Multiple Hereditary Exostoses
Article information

Multiple hereditary exostoses (MHE) is a disorder characterized by the growth of multiple osteochondromas in the metaphyses and diaphyses of long bones [1]. MHE is inherited in an autosomal dominant manner. The two causative genes of MHE, exostosin glycosyltransferase 1 (EXT1) and EXT2, are organized as a complex and are both required for heparan sulfate (HS) synthesis. Heterozygous mutations of either EXT gene result in HS deficiency [2]. EXT1 and EXT2 are known to cause exostoses by glycosyltransferases that are involved in the chain elongation step of HS biosynthesis, and HS influences important processes in skeletogenesis, skeletal growth, and morphogenesis [3]. A previous study reported that patients with deletion mutations including 8q24 involving EXT1 sometimes presented with autistic features [4]. HS regulates diverse cell-surface signaling events, and recent research has increasingly uncovered its roles in the development of the nervous system. Here, we describe the case of a boy with a microdeletion (8q24.11q24.13 [118,625,768-124,169,620]×1), who presented with autism, intellectual disability, and MHE. the parents signed informed consent and approved the anonymous use of clinical and molecular data for the present diagnostic study.
A boy aged 4 years and 10 months presented for an evaluation for autistic features and intellectual disability. He was the oldest child of unrelated parents with normal intelligence. He was born by cesarean section at term (41 weeks) after an uneventful pregnancy. His birth weight was 3,280 g (14th percentile). His mother was normal, and his father had a skeletal deformity around his knee; however, exact information was not available because the patient lived with his mother in a single-parent family. His younger sister was healthy with normal development. He had shown poor eye contact since infancy, and he could not construct full sentences and meaningful words. On physical examination, his height was 110 cm (60th percentile) and his weight was 15.4 kg (3rd percentile). He had dysmorphic features, including brachycephaly, a broad forehead, low-set ears, exotropia, and asymmetric and winged scapulae.
The patient showed global developmental delay on the Korean Bayley Scales of Infant Development-III; at 58 months old, his scores were as follows: cognitive scale, 19 months old; receptive communication scale, 17 months old; expressive communication scale, 4 months old; fine motor scale, 26 months old; and gross motor scale, 21 months old. The patient’s full scale intelligence quotient was 55 to 65, his social age was 3 years old, and his social quotient was 58. Brain magnetic resonance imaging and electroencephalography were normal. On computed tomography of the upper extremities, we observed multiple exostoses in the right proximal and distal radius and left proximal radius (Fig. 1). Tests for inborn errors of metabolism were normal. A single nucleotide polymorphism microarray detected a 5.5 MB microdeletion of 8q24.11q24.13 (118,625,768-124,169,620, hg19) encompassing EXT1, without trichorhinophalangeal syndrome type 1 (TRPS1) deletion (Fig. 2). The deleted region included 18 protein-coding genes (COL14A1, COLEC10, DEPTOR, DERL1, DSCC1, ENPP2, EXT1, HAS2, MAL2, MRPL13, MTBP, NOV, SAMD12, SNTB1, TAF2, TBC1D31, TNFRSF11B, and ZHX2), of which only EXT1 was confirmed to be related, with entries in the Developmental Disorders Genotype-to-Phenotype database [5].
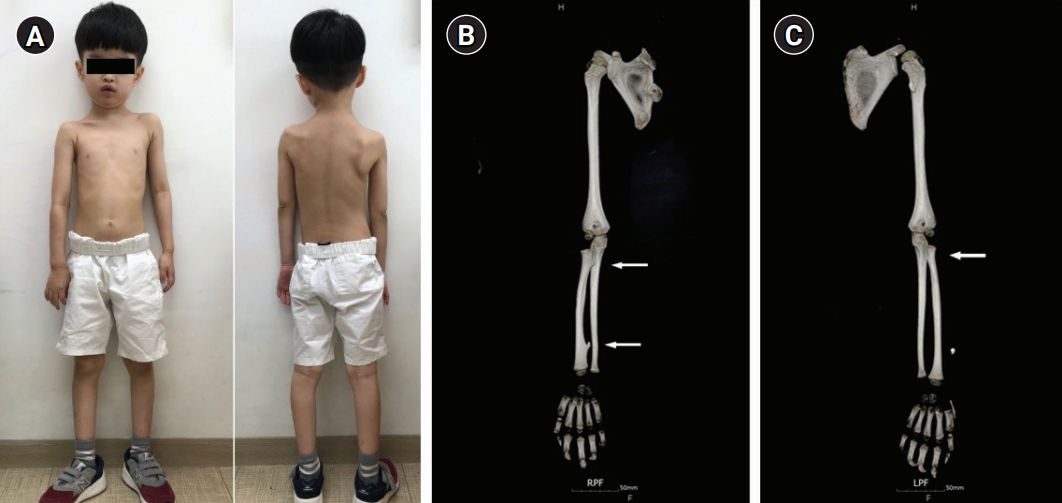
Asymmetry of the shoulder girdle with a higher position of the left side, scoliosis, and asymmetric winged scapulae (A) and computed tomography images showing exostosis in right proximal and distal radius (B) and the left proximal radius (C) (arrows).

Schematic representation of the 8q24 deletion and the contiguous gene syndromes, trichorhinophalangeal syndrome type 1 (TRPS1), Cornelia de Lange syndrome 4, and TRPS2. In this case, the 8q24.11q24 deletion encompassed exostosin glycosyltransferase 1 (EXT1) but not TRPS1.
The present case had a deletion in 8q24.11q24.13, including the EXT1 gene but not including the TRPS 1 gene. There are important contiguous genes in chromosome 8q24, such as TRPS1, RAD21, and EXT1. Deletions of those genes are associated with clinical syndromes such as TRPS1 (Online Mendelian Inheritance in Man [OMIM] #190350), Cornelia de Lange syndrome 4 (OMIM #614701), MHE involving EXT1 mutations, Langer-Giedion syndrome, and TRPS2 (OMIM #150230). The patient described herein had a 5.5 MB microdeletion (8q24.11q24.13) encompassing EXT1, but without TRPS1 deletion; therefore, he did not exhibit the typical features of Langer-Giedion syndrome such as cone-shaped epiphysis of the phalanges, delayed bone age prepubertally, and short stature.
In a previous study, MHE patients who had deletion mutations including 8q24 involving EXT1 were reported to have presented with autism [4]. EXT1 is expressed in the brain, and both the EXT1 and EXT2 proteins are associated with glycosyltransferase activities required for the biosynthesis of HS, which is also utilized in the brain [2]. This fact suggests that EXT1 may be involved in the development of mental disorders, including developmental delays and autism. HS regulates diverse cell surface signaling events. The roles of HS in neural development have been studied in animal models that carry mutations in EXT1, and these genetic studies revealed that HS is necessary for certain brain structures [3].
In conclusion, we describe a clinical association of autism, intellectual disability, and MHE in a boy with an 8q24.11q24.13 microdeletion encompassing EXT1, but not TRSP1. Based on this report and prior studies [3-5], the 8q24.11q24.13 microdeletion encompassing EXT1 is thought to be related to multiple exostoses and neurodevelopmental diseases, especially autism and intellectual disability.
This study was approved by the Institutional Review Board of Pusan National University Hospital (IRB, 2011-010-096). The requirement for written informed consent from the patient was waived due to the retrospective nature of our study.
Notes
Sang Ook Nam is an editorial board member of the journal, but he was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: YMK. Data curation: SON. Methodology: YL. Project administration: YL. Writing-original draft: MJK.