Heterogeneous Clinical Characteristics of Allan-Herndon-Dudley Syndrome with SLC16A2 Mutations
Article information

Abstract
Purpose
The purpose of this study was to expand our understanding of phenotypic and genetic variation in Allan-Herndon-Dudley syndrome (AHDS), which is a rare X-linked mental retardation syndrome characterized by hypotonia, generalized spasticity, and moderate-to-severe psychomotor retardation. AHDS is caused by a mutation of solute carrier family 16 member 2 (SLC16A2), which encodes monocarboxylate transporter 8 (MCT8), a transporter of triiodothyronine (T3) into neurons.
Methods
We enrolled nine patients with AHDS from unrelated families, except for two patients who were cousins, through a retrospective chart review. Clinical features, brain imaging, electroencephalograms, thyroid hormone profiles, and genetic data were reviewed retrospectively and compared with previously reported cases.
Results
We found three novel and five previously reported pathogenic variants in nine patients from eight families. All patients presented with hypotonia, spasticity, severe developmental delay, and elevated serum T3 levels. Cataplexy, which is a previously unreported phenotype, was found in two patients with the same mutation. In our cohort, seizures were uncommon (n=1) but intractable.
Conclusion
This study broadens the known phenotypic variations of AHDS, ranging from relatively mild global developmental delay to a severe form of encephalopathy with hypotonia, spasticity, and no acquisition of independent sitting. The syndromic classification or genetic etiology of global developmental delay is extremely heterogeneous; therefore, early clinical suspicion is challenging for clinicians. However, severe mental retardation with hypotonia, spasticity, and elevated serum T3 levels in boys is a highly suspicious clinical clue for the early diagnosis of AHDS.
Introduction
Allan-Herndon-Dudley syndrome (AHDS) is a rare disease characterized as an X-linked mental retardation syndrome that is associated with neuromuscular involvement and thyroid function abnormality. The first case of AHDS was reported by Allan et al. [1] in 1944, but its association with solute carrier family 16 member 2 (SLC16A2) mutation was first described in 2004 [2,3].
SLC16A2 encodes monocarboxylate transporter 8 (MCT8), a transmembrane transporter of the active form of thyroid hormone, triiodothyronine (T3) [4]. Thyroid hormones are important for neurodevelopment and normal growth. Because MCT8 is highly expressed in the brain–blood barrier and neuronal cells of the developing brain [5], MCT8 defects result in severe psychomotor deficits including central hypotonia, generalized spasticity, dystonia, and moderate-to-severe intellectual disability [2,3]. Impaired myelination on brain magnetic resonance imaging (MRI) has been reported in the majority of affected patients, but the heterogeneity of MRI findings needs further evaluation [6,7].
The thyroid profile of patients with AHDS shows elevated T3 levels, low-to-normal free tetraiodothyronine (T4) levels, and normal-to-high thyroid-stimulating hormone (TSH) serum levels [2,3]. Due to the elevated serum levels of T3, other organs, such as the heart, muscles, and liver, which are less dependent than the brain on the MCT8 protein for transporting thyroid hormones into the cell, are affected by peripheral thyrotoxicosis, the symptoms of which include muscle wasting, low body weight, and tachycardia [8,9].
The phenotypic spectrum of AHDS is highly varied, including patients who achieve independent walking with mild intellectual disability to those with severe encephalopathy [10,11]. The phenotype-genotype correlation is not yet understood fully, but residual protein activity is suggested to be associated with clinical severity [12].
Herein, we present nine patients who were diagnosed with genetically confirmed AHDS. Their clinical, laboratory, radiologic, and genetic characteristics were described in detail to broaden the scope of known phenotypic characteristics and to expand our understanding of the disease.
Materials and Methods
1. Patients
Nine male patients aged from 0 to 16 years were diagnosed with AHDS at Seoul National University Children’s Hospital from January 2010 to January 2020. The diagnoses were made by a clinical neurologist based on the patients’ clinical presentation and thyroid hormone profile followed by confirmative genetic testing. For the patients diagnosed with AHDS based on molecular analysis, we performed a detailed retrospective review of medical records including clinical characteristics, laboratory test results including the thyroid hormone profile, brain MRI, and electroencephalography (EEG).
2. Molecular analysis
Genomic DNA was extracted from peripheral blood from the patients and/or their family members by standard procedures after obtaining informed consent. Genetic variants were analyzed using targeted panels or whole-exome sequencing. The exon sequences and their flanking intronic sequences were identified through polymerase chain reaction using genomic DNA. The DNA sequences were mapped to and analyzed in comparison with the published human genome build (UCSC hg19 reference sequence [RefSeq]). The targeted coding exons and splice junctions of the known protein-coding RefSeq genes were assessed for the average depth of coverage and data quality threshold values. All reportable sequence variants below the threshold quality were confirmed by Sanger sequencing analysis using a separate DNA preparation. The DNA sequences were aligned to a human reference sequence (NCBI Accession No. NM_006517.5 for SLC16A2).
In silico analysis of the missense variants was performed using the CADD, SIFT, and Polyphen-2 programs, which evaluate the predicted deleteriousness and functional effects of each variant. The PhyloP program (phylogenetic P values) was used to measure evolutionary conservation. The minor allele frequency of variants was determined using data from gnomAD.
This study was approved by the Institutional Review Board (IRB) of the Seoul National University Hospital (IRB No.2101-173-1191). The requirement to obtain written informed consent from the patients was waived due to the retrospective nature of our study.
Results
1. Demographics and clinical presentation of AHDS syndrome
All nine patients were male and born from nonconsanguineous parents of Korean ethnicity. Table 1 summarizes the clinical manifestations and examination results of the nine patients who were genetically confirmed to have AHDS.
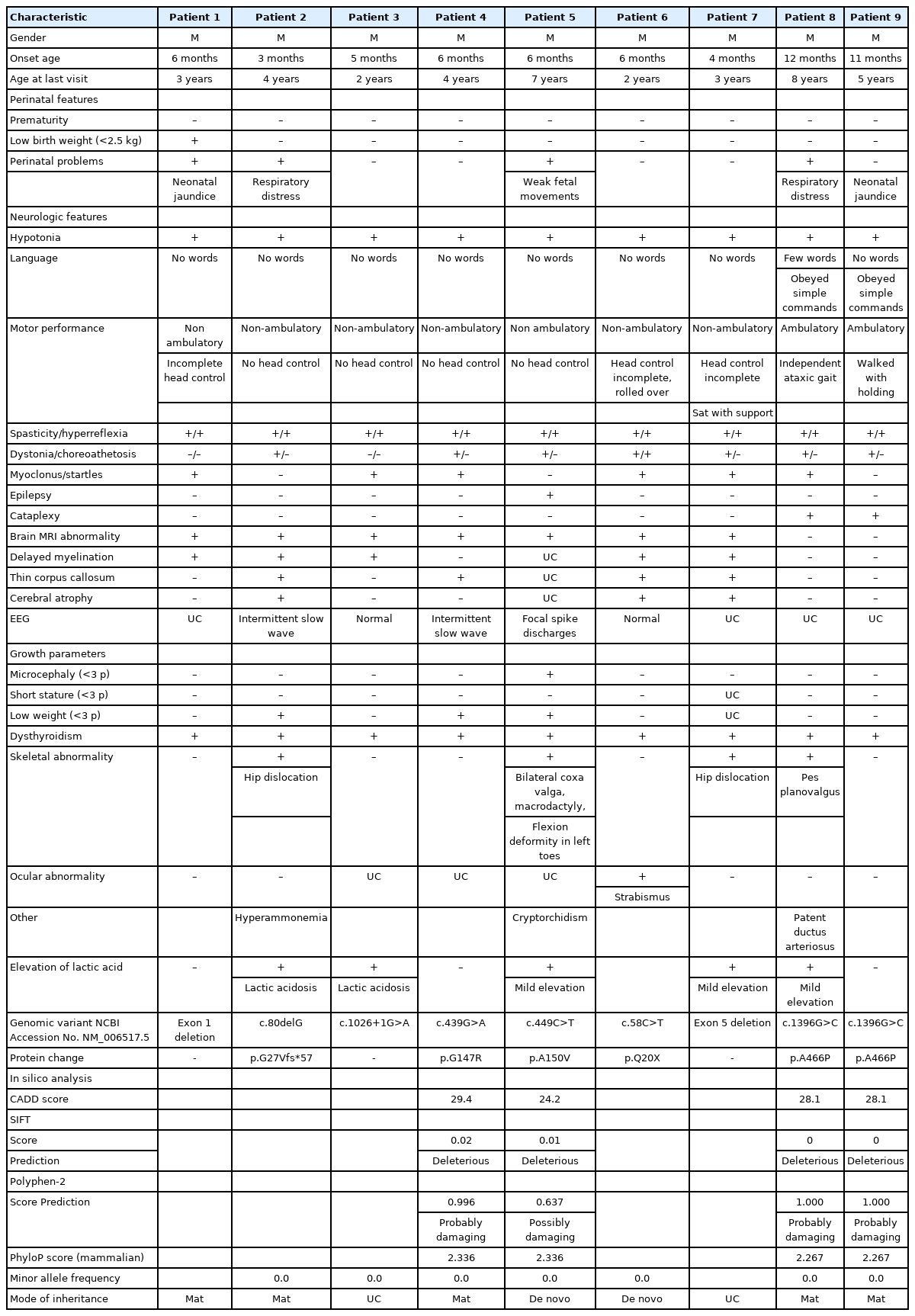
Phenotypic characteristics and genotypes of the nine patients with Allan-Herndon-Dudley syndrome
2. SCL16A2 mutation analysis and family history
We found three novel mutations (1396G>C [p.A466P], c.80delG [p.G27Vfs*57], c.58C>T [p.Q20X]), and five previously reported mutations (c.449C>T [p.A150V], c.439G>A [p.G147R], c.1026+1G>A, exon 1 deletion, exon 5 deletion) in nine patients from eight families. Five patients inherited the mutation from their mother and two patients had de novo mutations. In two patients, maternal carrier status was not checked. The mutations in the SLC16A2 are summarized in Table 1 and Fig. 1.
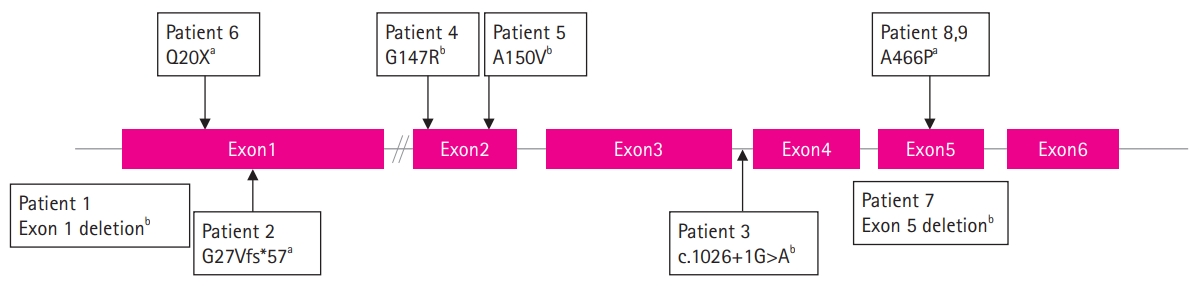
Mutations of the solute carrier family 16 member 2 (SLC16A2) gene in this study. aNovel variant; bPreviously reported variant.
Family histories were reviewed and relatives with cerebral palsy of unknown etiology were observed in three patients from two families. Patients 8 and 9 were maternal cousins and patient 3 had a maternal uncle bedridden with spastic paraplegia, who was considered to have cerebral palsy from an unknown cause.
3. Perinatal problem
Antenatal problems were reported in one of the nine patients. Weak fetal movement, a small biparietal diameter, and low body weight were observed in patient 5, but delivery was uneventful. All patients were born full-term. Neonatal problems were present in three patients (33.3%). The patients with neonatal jaundice (n=2) were treated by phototherapy, those with transient respiratory distress (n=2) did not need ventilator support, and one patient with patent ductus arteriosus (n=1) required a ligation operation. Patient 8 presented with neonatal jaundice, transient respiratory distress, and patent ductus arteriosus; however, patient 9, who had the same mutational variant as patient 8, did not experience any of these problems.
4. Growth
The growth parameters at birth were all normal for all patients except patient 1, who had low birth weight. Although all patients except one had a normal body weight at birth, poor weight gain was present in three patients. Microcephaly was present in patient 5, who also had low body weight (below the first percentile). Short stature was not observed in any of the nine patients during the follow-up period.
5. Neurologic symptoms
All patients visited the clinic for evaluations of motor developmental delay. The median age at the first visit was 6 months (range, 3 to 12) and the median age at the last follow-up was 4 years (range, 2 to 8). In seven patients, the initial symptom was an inability to control the head at the age of 3 to 6 months. In patients 8 and 9, who were maternal cousins with the same variant, developmental delay was noticed at 11 to 12 months of age because they could not sit alone by then. These two patients could walk with a dystonic and ataxic gait at 4 years old. The other seven patients who could not walk independently showed various patterns of motor development during the follow-up period, as follows: no head control (n=2), partial head control (n=3), partial head control and rolling over (n=1), and partial head control and sitting with assistance (n=1). Axial hypotonia was observed in all patients (n=9). Pyramidal symptoms were also present in all patients, including spasticity (n=9) and hyperreflexia (n=9). Extrapyramidal symptoms were also frequent, including dystonia (n=7) and choreoathetosis (n=1). Notably, patient 9 showed no definite abnormality on the initial neurologic examination at the age of 11 months, and did not demonstrate axial hypotonia at that time. Other pyramidal and extrapyramidal features became evident and progressed over time.
Startles or myoclonic movements were common symptoms (n=5). This symptom was triggered especially when the patients were about to sleep or were stimulated by sounds. Four patients underwent an EEG for startles or myoclonic movements to differentiate them from seizures. The EEG records from two patients were normal and the other two patients showed intermittent high-amplitude delta activities without definite epileptic discharges.
Seizures were observed in one patient. Specifically, seizures started at 3 months of age in patient 5, who was intractable to multiple antiepileptic drugs. The seizure semiology was generalized as tonic or tonic-clonic, and seizures occurred several times a day. EEG studies revealed focal epileptic discharges in the unilateral frontocentral region.
All patients had an intellectual disability, but its severity varied. Seven patients never walked independently or spoken a single word; however, they could make eye contact and social smiles, and recognize their parents. Patient 8 could say a few words, but was not able to make a sentence and could comprehend simple commands. Patient 9 who had the same mutation variant as patient 8 could not say any words, but could communicate with gestures and understand simple commands. Interestingly, patient 8 showed regression of language ability at the age of 7 years in both expressive and receptive language.
Cataplexy, which is a sudden loss of muscle tone triggered by emotional stress, was observed in patients 8 and 9 at the ages of 7 and 5 years, respectively. No other patients in this cohort showed cataplexy.
6. Brain MRI
Among the nine patients, patients 8 and 9, who had the same SLC16A2 mutation variant, showed normal brain MRI findings on examinations performed at 1, 2, and 8 years and 1 year of age, respectively. Abnormalities on brain MRI were observed in seven patients, including delayed myelination, brain atrophy, a thin corpus callosum, and a prominent subarachnoid space in the middle cranial fossa. Delayed myelination was found in five patients. Among them, serial MRI studies were obtained in one patient and delayed myelination was persistent until the last follow-up at 4 years of age (Fig. 2). Diffuse cerebral atrophy was found in five patients, and serial MRI studies in two of them revealed progression of cerebral atrophy. A thin corpus callosum was found in four patients. In one of them, a thin corpus callosum was the only abnormality observed on brain MRI. A subarachnoid cyst or prominent subarachnoid space in both middle cranial fossae was found in two patients.
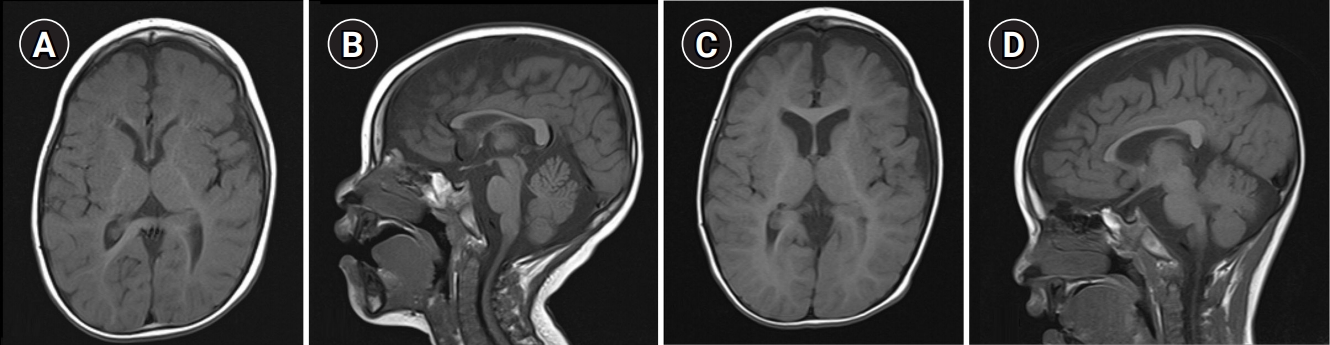
Brain magnetic resonance imaging (MRI) of patient 2 at the ages of (A, B) 22 months and (C, D) 4 years. (A, C) Axial and (B, D) sagittal T1-weighted MRI showed a thin corpus callosum, progression of cerebral atrophy, and persistent hypomyelination.
7. Thyroid hormone blood levels
The thyroid hormone profiles at patients’ initial examinations are summarized in Table 2. The median age at the first test was 16 months (range, 8 to 83). TSH levels were normal in eight patients and high in patient 1. Patient 1 had low free T4 with a high TSH level; therefore, he was treated with levothyroxine replacement before the diagnosis of AHDS. Low free T4 levels were found in four patients and were normal in five patients. T3 levels were high in all nine patients at the initial test. The median T3 level at the initial test was 250 ng/dL (range, 190 to 436.5; reference, 64 to 152). In patient 5, successive thyroid hormone tests revealed normalization of T3 levels and decreased free T4 levels below the normal range at 4 and 5 years of age compared to the initial results obtained at 9 months of age. The ratio of T3 to free T4 was also calculated, and the median T3 to free T4 ratio was 420.7 (range, 231.7 to 704.0; reference, 41.2 to 217.1). The T3 to free T4 ratio was consistently high even in patient 5, whose T3 level was normalized and had a decreased free T4 level.
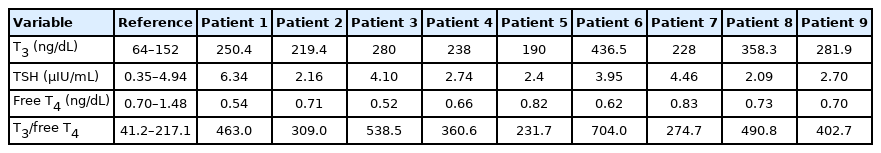
Initial thyroid hormone levels
8. Laboratory abnormalities
Elevated lactic acid levels were observed in five patients. Severe lactic acidosis needing alkali treatment was observed in two patients and hyperlactatemia without metabolic acidosis was observed in the remaining three patients. Transient hyperammonemia accompanied by severe lactic acidosis was observed in patient 2, which was incidentally identified from the metabolic workup and treated with sodium benzoate. Neurologic symptoms associated with hyperammonemia were not observed. After that event, the ammonia level remained in a normal range even after ceasing medication. Intermittent elevation of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and creatinine kinase (CK) was noted in two patients.
9. Skeletal problems and other problems (urologic and ocular)
Skeletal abnormalities were found in four patients, including hip dislocation (n=2), bilateral coxa valga (n=1), macrodactyly of the first and second toes (n=1), and pes planovalgus (n=1). Cryptorchism was present in patient 5 and testis sonography revealed unilateral gliding testis. Ocular examinations were performed in six patients, and strabismus was noted in one patient.
Discussion
In this study, we conducted a detailed investigation of the clinical, genotypic, and laboratory characteristics of nine male patients with genetically confirmed AHDS.
After the causative gene SLC16A2 was revealed, the pathophysiology of AHDS has been understood as resistance to thyroid hormones in the brain due to the deficiency of the thyroid hormone transporter, MCT8, which plays a key role in transporting thyroid hormones across the blood–brain barrier and into neuronal and glial cells in developing brains [13]. The MCT family, which is a member of the major facilitator superfamily (MFS), has a shared structural fold with 12 transmembrane domains (TMDs) and cytosolic N- and C-termini [14]. This transmembrane protein is thought to control the cellular influx and efflux of thyroid hormones and regulate intracellular hormone levels [4,15]. An expression analysis of MCT8 revealed that it is present in cerebral neurons, glial cells, and the cellular structures that compose the blood–brain barrier and blood–cerebrospinal fluid barrier in the fetal brain [5,16]. In addition to the brain, MCT8 is also abundant in the thyroid glands, kidneys, and liver [4].
A distinctive thyroid hormone profile is a hallmark of AHDS [2,3]. Typically, the T3 level is elevated, the free T4 level is normal to low, and the TSH level is normal to mildly elevated. Considering the diseases resulting from defects of other thyroid hormone transporters, such as OATP1C1 deficiency, which shows a normal thyroid hormone profile [17], abnormal serum thyroid hormone levels not only provide a clue for diagnosis, but also show the association of MCT8 and the hypothalamus-pituitary-thyroid (HPT) axis. Studies on the relevance of MCT8 to the HPT axis in Mct8-knock-out mice have shown that the hypothalamus and pituitary gland seem to poorly sense serum thyroid hormone concentrations, which suggests that TSH level is not a reliable indicator of the thyroid hormone status of the brain and body [18,19]. Although an elevated T3 level was thought to be a fingerprint of AHDS, normal T3 levels were reported in some cases [10,11]. In our cohort, patient 5, who had an elevated T3 level of 190 ng/dL (reference range, 64 to 152) at the age of 9 months showed normal serum T3 levels of 144.5 and 115.7 ng/dL at the ages of 4 and 5 years, respectively, with decreased free T4 levels, but the ratio of T3 to free T4 was still elevated. If AHDS is clinically suspected, it is recommended to test the patients’ thyroid hormone status repeatedly and keep in mind that the T3 level can vary. An increased T3 to free T4 ratio may be a more reliable parameter for clinical suspicion of AHDS.
The types of mutations in the SLC16A2 gene vary considerably, from single-nucleotide variants to large deletions. In our cohort, we reported three missense mutation, one nonsense mutation, one frameshift mutation, one splicing-site mutation, and two exon deletions. Several functional studies have shown that genetic mutations of SLC16A2 cause a variable degree of functional defects in MCT8, including impairments of homodimerization, localization to the plasma membrane, and transport function [20,21]. Two previously reported missense mutations were found in patients 4 and 5, who had c.439G>A [p.G147R] and c.449C>T [p.A150V] mutations, respectively. The c.439G>A [p.G147R] mutation changes the glycine located in the second TMD to arginine. In functional studies [20], it impaired the thyroid hormone transporter activity without affecting localization to the plasma membrane. The clinical manifestation of two patients with the p.G147R mutation from the same family reported previously [18] was similar to patient 4, who had a severe phenotype with spastic paraplegia and absence of verbal output, except for seizures, which were not observed as of the age of 4 years (at his last visit). The c.449C>T [p.A150V] mutation in patient 5 changed alanine to valine in the second TMD. This has been reported previously, and all cases had the same severe phenotype as patient 5 [19,22,23]. In the functional analysis [19,21], this mutation was reported as not affecting dimer assembly [21], but impairing expression in the plasma membrane [19]. The novel 1396G>C [p.A466P] missense mutation from patients 8 and 9 changed alanine to proline in the 11th TMD and caused a relatively mild phenotype, as the patients gained the ability to walk and had normal brain MRI findings. A functional study was not performed for these patients. In our cohort, exon deletion was found in patients 1 and 7, who showed exon 1 and exon 5 deletion, respectively; sole exon 5 deletion has not been reported previously. Both patients had a severe phenotype, with spastic paraplegia and no verbal output. Patient 3 had a c.1026+1G>A spicing site mutation, which was previously reported in one case [24]. In that case, the clinical presentation was similar, showing a severe phenotype, but the thyroid hormone profile was different because the previously reported case showed lower free T4 levels than patient 3, who had normal free T4 levels [24]. The novel frameshift mutation c.80delG [p.G27Vfs*57] and nonsense mutation c.58C>T [p.Q20X] on exon 1 were observed in patients 2 and 6, respectively, who had severe phenotypes.
Axial hypotonia and spasticity were the most common features in our cohort, which corresponds well with previously reported cases [10,11]. Among several extrapyramidal phenotypes, exaggerated startles or myoclonus was a notable phenotype in this cohort, as observed in 55% (5/9) of the patients. Dystonia, which is also a commonly reported manifestation of AHDS, was also widely observed in this study.
Along with the increasing number of reported patients and genetic mutations of AHDS over the last decade, some patient groups have shown milder phenotypes than others, with mild intellectual disability or achieving ambulation [10,18,25-29]. In this cohort, patients 8 and 9, who had the same missense 1396G>C [p.A466P] mutation at the end of the 11th TMD, showed a distinctively milder phenotype compared to the other patients. Their age of onset was later than that of the others and they achieved ambulation around 5 years of age. Although both had the same variants and could be categorized as having a mild phenotype, they had phenotypic differences. At the initial neurologic examination at the age of 12 months, patient 9 showed more classical clinical features, such as hypotonia, spasticity, and hyperreflexia, whereas patient 8 showed none of these features except for mild hypotonia. During the follow-up period, these clinical features became evident in patient 8. In this patient with a mild phenotype, the initial neurologic examination was uncertain; thus, thoughtful follow-up is crucial.
To our knowledge, cataplexy, which was a notable shared phenotype of patients 8 and 9, has not previously been reported in patients with AHDS. As one of the features of narcolepsy, cataplexy is thought to be related to the deficiency of orexin, a neurotransmitter secreted by neurons in the lateral hypothalamus [30], which plays a role in the sleep–wakefulness cycle. There is a topographical similarity in that both orexin-producing neurons and MCT8-expressing cells exist in the hypothalamus [31]. Although the underlying mechanism is unclear, several studies have shown evidence supporting the association between the HPT axis and the orexin system. In an animal study, treatment with a thyrotropin-releasing hormone analogue improved cataplexy [32] and a low level of TSH was reported in patients with orexin-deficient narcolepsy [33]. The finding of cataplexy in patients with AHDS could also support the suggested link between the two systems; thus, further research is needed.
Several phenotypes in patients with AHDS, including poor weight gain, decreased muscle bulk, and tachycardia as reported previously, were thought to be caused by peripheral thyrotoxicosis. Poor weight gain was also a common phenotype in this cohort. Skeletal muscle is one of the major target organs for thyroid hormones, which regulate homeostasis. The regeneration of skeletal muscle and excessive T3 levels in patients with AHDS changes the expression pattern of numerous proteins toward a catabolic status, which results in muscle wasting [34]. Elevation of lactic acid was observed in about half of the patients, and one-third of them experienced metabolic acidosis. Along with lactic acid, the increased levels of AST, ALT, CK, and ammonia were thought to reflect the catabolic status of skeletal muscle [8]. These laboratory abnormalities can sometimes lead to the misdiagnosis of mitochondrial or muscle disease; for example, previous reports of brain MRI findings in patients with AHDS [6] showed that delayed myelination or hypomyelination was a major accompanying feature with or without cerebral atrophy. In some previous research, normalization of myelination in the successive follow-up of brain MRI was reported [6,7]; however, this was not observed in our case. The observation of severe hypomyelination on brain imaging can lead to a misdiagnosis of other neurodegenerative diseases that are categorized into leukodystrophy, such as lysosomal storage disease or mitochondrial disease. Normal brain MRI findings were also observed in two patients with milder phenotypes, which makes AHDS difficult to diagnose.
Due to the specific pathophysiology and significant burden of AHDS, various efforts have been made to obtain an effective treatment [35]. Recently, a second-phase clinical trial of treatment with triiodothyroacetic acid, which is a thyroid hormone analogue, showed clinical improvement of peripheral thyrotoxicosis and the possibility of improving patients’ neurologic status if treated at a young age [36]. Therefore, an early diagnosis is becoming more important.
Herein, we report nine patients who were genetically confirmed with AHDS with mutations of SLC16A2, which encodes MCT8, a thyroid hormone transporter. Phenotypic and genetic variability is a notable feature of the disease, which makes an early diagnosis difficult. Understanding its broad clinical features, which reflect the opposite patterns of thyroid hormone status in the brain and periphery, with a variable serum thyroid profile and MRI findings will help to stimulate clinical suspicion and guide early confirmatory genetic tests. Importantly, a thyroid function test including T3 levels should be considered when encountering male patients with severe developmental delay and spasticity.
Notes
Jieun Choi is an associate editor, Ki Joong Kim and Jong-Hee Chae are the editorial board members of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: SYK, HK, BCL, HH, JC, and JHC. Data curation: JYH, SL, and HW. Formal analysis: JYH and SL. Funding acquisition: JHC. Methodology: JYH, HW, HH, and JHC. Project administration: SYK, HK, KJK, and JHC. Visualization: JYH. Writing-original draft: JYH. Writing-review & editing: BCL, KJK, and JHC.
Acknowledgements
This research was supported by the Seoul National University, Department of Pediatrics and Rare Disease Center.