X-linked ichthyosis (XLI; MIM #308100) is a dermatologic disorder characterized by large, dark brown scales similar to fish scales. It starts primarily on the extensor surfaces and the side of the trunk symmetrically, becomes widely distributed, and then disappears throughout childhood. Most cases of XLI are caused by mutations in the steroid sulfatase (STS) gene (online Mendelian inheritance in men [OMIM] * 300747), which is located on the short arm of the X chromosome at Xp22.3 and encodes the STS enzyme [1]. Most XLI patients (90%) have large deletions within or including STS [2]. These patients display various clinical symptoms, as well as skin lesions. Baek and Aypar [3] reported a 5-year-old girl who presented with mild autism, attention-deficit hyperactivity disorder (ADHD), and dry, scaly skin on the body. A chromosomal microarray (CMA) revealed a large deletion, including STS and neuroligin 4, OMIM*300427 (NLGN4), associated with ADHD and autism [3]. Here, we report three children with XLI and developmental delay. We confirmed XLI and the presence of a large deletion through CMA with a single-nucleotide polymorphism (SNP) array and also investigated other associated clinical symptoms.
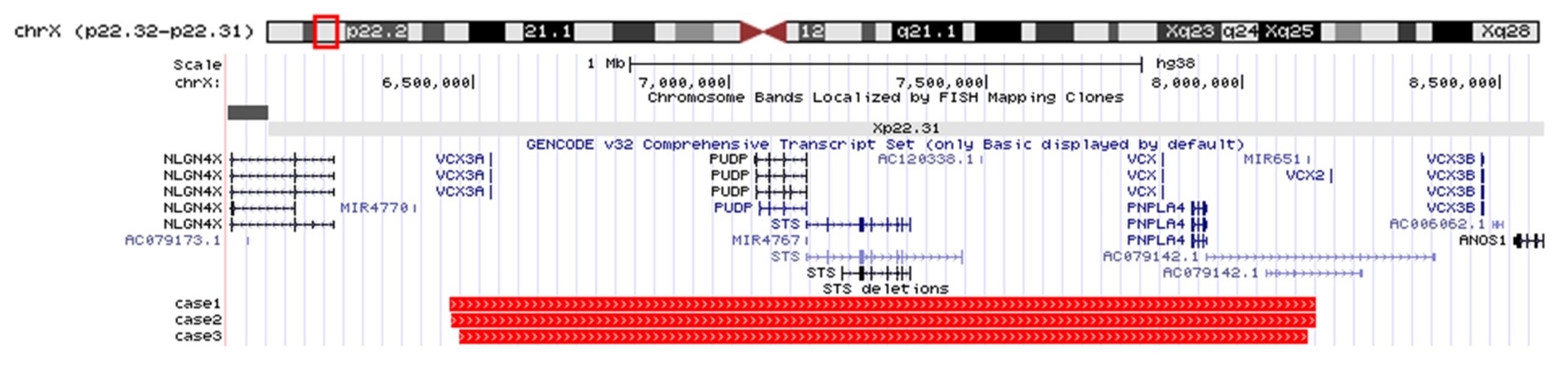
The clinical characteristics of the three patients and their CMA results are summarized in Table 1. Patient 1 was a 7-year-old boy who was born at 35 weeks 6 days by cesarean section due to maternal preeclampsia. His birth weight was 1,886 g and he showed intrauterine growth retardation. He underwent surgery due to coarctation of aorta on day 6 after birth. At 12 months of age, he presented with a nevus on his forehead, dry and scaly skin, increased muscle tone, and global developmental delay. No family members had the typical skin lesions of XLI. Magnetic resonance imaging (MRI) of the brain and an electroencephalogram (EEG) were normal. The karyotype was normal. The patient received continued rehabilitation, but the developmental delay did not improve appreciably. He showed no sign of autism or ADHD. His full-scale intelligence quotient (FSIQ) was 35 to 40 and social quotient (SQ) was 37. CMA was performed with a CytoScan 750K array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s recommendations in all three patients. The SNP array revealed a deletion of approximately 1.7 Mb at Xp22.31 (6,455,151 8,141,076). The deleted region included several genes, such as variably charged, X chromosome, 3A, OMIM*300533 (VCX3A), haloacid dehalogenase-like hydrolase domain-containing 1A, OMIM*306480 (HDHD1A), STS, VCX (OMIM * 300229), and patatin-like phospholipase domain-containing protein 4, OMIM*300102 (PNPLA4). The patient was diagnosed as having XLI with intellectual disability. At 9 years of age, he presented with general tonic seizures and focal interictal discharge over the occipital area on EEG at the onset of the seizure. We diagnosed the patient with epilepsy. The patient is now 11 years of age and has epilepsy, myopia, and scoliosis. He cannot speak any words and cannot sit or walk alone.
Patient 2 was a 21-month-old boy with dry and scaly skin since birth. He was born at term by cesarean section. His birth weight was 2,460 g and he showed intrauterine growth retardation. His maternal grandfather and a maternal cousin had ichthyosis. His parents noticed his global developmental delay at approximately 12 months of age. In the Korean Infant and Child Developmental Test, he showed global developmental delay, with a developmental age (DA) of gross motor skills of 12 months old (developmental quotient [DQ] 57, normal DQ >85), a DA of fine motor skills of 16 months old (DQ 76), a DA of social-personal development of 12 months old (DQ 57), a DA of language development of 16 months old (DQ 76), and a DA of cognitive function of 16 months old (DQ 76). Brain MRI and an EEG scan were normal. Tests for inborn errors of metabolism were normal. He showed no signs of autism or ADHD. The SNP array revealed a deletion of approximately 1.7 Mb at Xp22.31 (6,473,896-8,127,579), which included VCX3A, HDHD1A, STS, VCX, and PNPLA4. He was diagnosed with XLI and global developmental delay. At the time of this report, the patient cannot walk alone and speak any significant words, but his height and weight are in the normal range.
Patient 3 was a 3-year-old girl born at term. She showed axial hypotonia, dry skin without scales, and global developmental delay during infancy. She underwent surgery for strabismus at 2 years of age. There was no family history of ichthyosis. Brain MRI and an EEG scan were normal. Tests for inborn errors of metabolism were normal. She showed no signs of autism or ADHD. Her FSIQ was 55 and SQ was 56. The SNP array revealed a deletion of approximately 1.7 Mb at Xp22.31 (6,458,939-8,143,509), which included VCX3A, HDHD1A, STS, VCX, and PNPLA4. She was diagnosed as being a carrier of XLI with a milder skin phenotype than that of male XLI, intellectual disability, and strabismus. She can now walk alone, but runs imperfectly. Her height and weight are in the normal range.
XLI is an X-linked recessive disorder that occurs almost exclusively in males. It has been rarely reported in homozygous women who were offspring of an affected man and a female carrier [4]. Approximately 90% of patients with XLI have a large deletion that includes STS. The remaining cases are caused by point mutations within STS. The encoded STS protein belongs to the sulfatase family. The enzyme hydrolyzes several 3-beta-hydroxysteroid sulfates, and specifically converts the sulfated form of dehydroepiandrosterone (DHEA), otherwise known as DHEAS, to DHEA. In rodents, increased blood levels of DHEAS and DHEA can result in enhanced memory [5]. In humans, lower levels of DHEA in the blood have been reported to be associated with diverse phenotypes, such as ADHD, corneal opacities, cryptorchidism, epilepsy, and developmental delay [1]. Patients with STS escape from X-inactivation (lyonization) and heterozygous carriers have been reported to exhibit increased rates of neurodevelopmental disorders, elevated levels of inattention, impulsivity, autism-related traits, and general psychological distress [6]. Placental STS deficiency can occur during pregnancy in XLI fetuses of a carrier mother, and the risk of obstetric complications, such as delayed and prolonged labor, is higher than in the general population [1]. In the present report, patient 3 was a girl with a heterozygous deletion of Xp22.31 including STS. Although she displayed global developmental delay and intellectual disability, she showed a mild skin phenotype with only dry skin, not the typical scaly skin of XLI.
We present three cases of XLI with a deletion of approximately 1.7 Mb at Xp22.31. All three patients presented with dry, scaly skin and developmental delay. The SNP array revealed large deletions that included STS, HDHD1A, VCX3A, VCX, and PNPLA4 (Fig. 1). HDHD1A encodes a member of the haloacid dehalogenase-like (HAD) hydrolase superfamily. The encoded protein has no known biological function. PNPLA4 encodes a member of the patatin-like family of phospholipases. The encoded enzyme has triacylglycerol lipase and transacylase activities, and it may be involved in adipocyte triglyceride homeostasis. It has been reported that PNPLA4 might be associated with autism [7]. Variable charge, X-linked (VCX) belongs to the VCX/Y family, which has multiple members on both the X and Y chromosome, and all are expressed exclusively in male germ cells. VCX/Y genes encode small and highly charged proteins of unknown function. VCX3A, also known VCXA, has been reported to show an association with abnormal neurologic phenotypes. Fukami et al. [8] showed that VCXA was preserved in all XLI patients without developmental delay, whereas it was deleted in all XLI patients with developmental delay. Hosomi et al. [9] identified a VCX3A promoter deletion in a case of XLI with borderline mental retardation. The collective findings indicate that deletions of VCX3A or STS might be associated with developmental delay in the XLI patients described in this report. We can assume that patients who have deletions of both VCX3A and STS show a greater variety of phenotypes and more severe neurologic symptoms.
In conclusion, the three present cases, in combination with previous studies, indicate that XLI can be associated with developmental delay. Cytogenetic tests are an important tool to identify and predict the risk of neurologic manifestations. We believe that our report will improve awareness of the neurologic manifestations of XLI on the basis of cytogenetic tests.
This study was approved by the Institutional Review Board of Pusan National University Hospital (approval number: 2007-030-093). Written informed consent was obtained from the parents of all patients.