Striatal necrosis (SN) has a unique clinical presentation with characteristic neuroradiological findings. It has mainly been noted in infancy with developmental arrest, dystonia, choreoathetosis, spastic quadriparesis, and optic atrophy [1]. We report an Indian family of five siblings with dystonia, intellectual disability, optic atrophy, and SN on brain magnetic resonance imaging (MRI), with symptoms occurring from the age of 6 to 10 years.
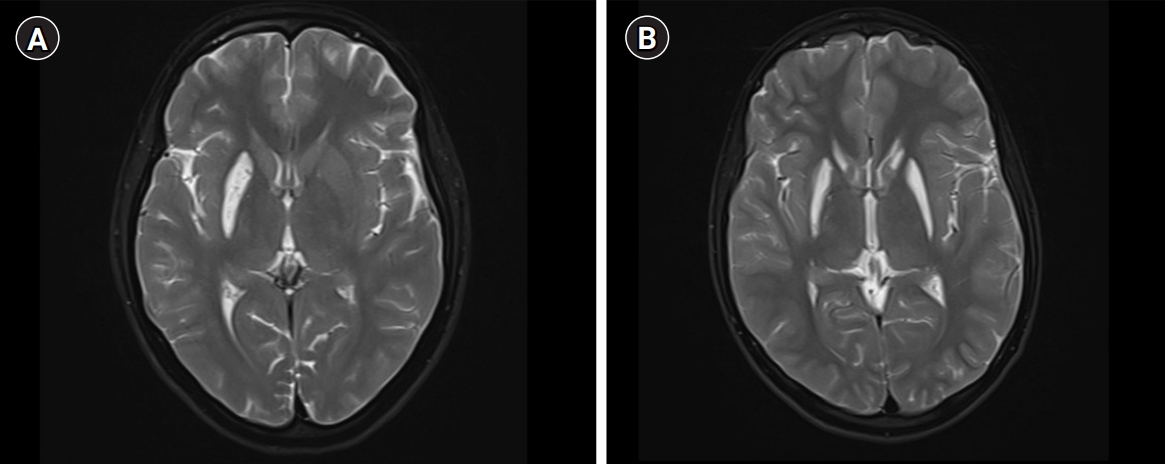
A 17-year-old girl presented with insidious-onset and gradually progressive dystonic posturing of the left upper limb of 6 years’ duration. It was initially present during movement or when performing tasks and later was present even at rest. She developed torticollis a year later. Her birth history and development were normal. She had a significant family history of similar dystonia of the limbs in four of her six siblings, with the proband being the second-born in a family of seven children born to non-consanguineous parents (Fig. 1). There was no history of movement disorders or intellectual disability in the other generations. On examination, she had head circumference of 52 centimeters, subnormal intelligence, hypometric saccades and impaired vertical optokinetic nystagmus (OKN), bilateral optic atrophy, and left upper limb dystonia with compensatory biceps hypertrophy. She had torticollis, facial grimacing, and left foot dystonia on walking. Rigidity was noted in the left upper limb and bilateral lower limbs with bradykinesia in the left upper limb. The deep tendon reflexes were normal and plantar responses were equivocal. She had elevated serum ammonia and elevation of alanine on tandem mass spectrometry screening of the blood for amino acids and acyl carnitine. Blood lactate and serum copper and ceruloplasmin levels were normal. Brain MRI revealed T2 hyperintensity, T1 hypointensity, and fluid-attenuated inversion recovery hyperintensity mixed with areas of hypointensity in the right putamen suggestive of SN with no evidence of involvement of the subthalamic nuclei, brainstem, cerebellum, or white matter (Fig. 2). Magnetic resonance spectroscopy (MRS) of the lesion did not show any abnormalities. Muscle biopsy of the left biceps did not reveal any abnormality on histopathology and enzyme histochemistry. Genetic testing for mitochondrial Leigh’s disease mutations by bidirectional Sanger sequence analysis (T12706C, A13084T, and G13513A) was negative.
A detailed clinical and radiological evaluation of her affected siblings was conducted. The third-born child, a 15-year-old boy, had left hemidystonia and torticollis with onset from the age of 7 years. He had a head circumference of 52 cm, mental subnormality, bilateral optic atrophy, and impaired vertical OKN. He had rigidity of the left upper limb and bilateral lower limbs, bradykinesia, and bilateral extensor plantar response. The fourth-born, a 13-year-old female child, was the most severely affected; she had tetraplegic dystonia, with onset from age of 7 years. She had a head circumference of 50 cm, short stature, mental subnormality, and normal fundus and visual acuity with blepharospasm. A motor examination revealed bradykinesia, rigidity of all four limbs, and extensor plantar response. The fifth born, a 10-year-old female child, had dystonia of the right upper limb and bilateral lower limbs with onset from the age of 7 years. She had a head circumference of 51 cm, mental subnormality, pale optic discs, impaired vertical OKN, and a staring look with bilateral upper eyelid retraction. Rigidity and bradykinesia with extensor plantar response were noted on examination. The sixth born, an 8-year-old female child, had onset of right lower limb and left upper limb dystonia from age of 6 years. She had a head circumference of 50 cm, was mentally subnormal, had bilateral pale discs, impaired vertical OKN, and generalized rigidity with a bilateral striatal toe. Brain MRI of all the affected siblings revealed bilateral SN (Fig. 2). However, MRS and detailed blood or cerebrospinal fluid investigations in the siblings were not done. With further genetic evaluation pending due to financial constraints, the children were empirically initiated on symptomatic treatment. A trial of levodopa in the proband did not lead to any improvement. A mitochondrial etiology was considered possible in view of the clinical features of optic atrophy, intellectual disability, pyramidal and extrapyramidal symptoms, and biochemical changes such as elevated alanine, and the children were started on a mitochondrial cocktail. They were given 20 mg/kg/day of ubiquinone, 100 mg of riboflavin, 50 mg/kg/day of carnitine, 100 IU of vitamin E, 100 mg of vitamin C and 100 mg of alpha-lipoic acid daily. Biotin at a dose of 5 mg/kg/day and thiamine at a dose of 30 mg/kg/day were also initiated and the children were followed up. However, no clinical improvement was noted on follow-up after 6 months.
SN is histopathologically defined by swelling of the putamina and caudates followed by degeneration and cellular necrosis [2]. Radiologically, swelling followed by striatal atrophy and cavitation is noted. SN may be seen in the following conditions: Leigh’s disease; organic acidurias such as glutaric aciduria type I, methyl malonic acidemia, propionic aciduria, and isovaleric aciduria; biotin thiamine-responsive basal ganglia disease; thiamine-responsive basal ganglia disease; nucleoporin 62 (NUP62)-related disorder; and rarely, after infections caused by Mycoplasma pneumoniae, human herpesvirus 6, and herpes simplex virus 1 [3]. In the affected family, the proband had milder unilateral symptoms than her siblings, a later age of onset, and unilateral SN. The age of onset of dystonia was 6 to 11 years of age in the family, unlike the more common infantile presentation, and the disease had a much slower progression and a less devastating course. The salient clinical features were intellectual disability in all five of the affected children, bilateral optic atrophy, eye movement abnormalities such as hypometric saccades and impaired OKN, appendicular rigidity, generalized dystonia, and extensor plantar response. Mutations in the NUP62 and adenosine deaminase acting on RNA 1 (ADAR1) genes have been noted in infantile familial bilateral SN [4,5]. A few case reports mention mutations in the mitochondrial gene reduced nicotinamide adenine dincleotide dehydrogenase subunit 6 (ND6) responsible for a similar onset in childhood and in the phosphodiesterase 8B (PDE8B) gene in adults [6-8].
Mitochondrial disorders such as Leigh’s disease, Leber hereditary optic neuropathy with dystonia and mitochondrial encephalomyopathy, and lactic acidosis and stroke‐like episodes (MELAS) can be associated with SN. A comprehensive genetic evaluation including whole exome sequencing with mitochondrial genome sequencing would be necessary in this family. A detailed genetic evaluation of the above family is pending and we intend to report the results of the analysis in a future publication. SN may have a late age of onset, unlike what has been previously known, and this report adds to knowledge on the clinical spectrum of the disease and the relevant literature from India.
Informed consent was waived according to institutional regulations.