Mega-corpus callosum is a rare radiological finding. It is a characteristic finding of diseases such as Cohen syndrome, neurofibromatosis (NF), megalencephaly-polymicrogyria-mega-corpus callosum (MEG-PMG-MegaCC, MIM#603387) syndrome or mega-corpus callosum syndrome with cortical malformations (MCC-CM, MIM#618273) [1]. Such diseases are characterized by specific phenotypes such as neurofibromas in NF; non-progressive mental retardation and microcephaly in Cohen syndrome; megalocephaly and polymicrogyria in MEG-PMG-MegaCC; and normocephaly and cortical malformation, with or without cerebellar hypoplasia in MCC-CM. The knowledge of these specific phenotypes could facilitate the differentiation of these conditions from each other. However, MCC-CM has been reported to be a phenotype of MEG-PMG-MegaCC [1]. The detection of pathogenic variants in the microtubule associated serine/threonine kinase 1 (MAST1, MIM*612256) gene, which plays a role in human brain development and its relationship with neurodevelopmental phenotypes, has distinguished MCC-CM as a condition distinct from MEG-PMG-MegaCC [2]. MCC-CM, which is caused by autosomal dominant heterozygous mutations in the MAST1 gene, has been reported in less than 10 patients to date. Herein, we describe the first case of a male patient with MCC-CM with cerebellar hypoplasia in Korea. The diagnosis was based on the detection of MAST1 mutation via whole exome sequencing (WES). The patientŌĆÖs other manifestations such as microcephaly and lack of secondary sexual characteristics have never been reported thus far. This study was approved by the Institutional Review Board of Inje University Busan Paik Hospital (18-0041), and the data were presented without divulging the patientŌĆÖs identity. Written informed consent was obtained from a patient.
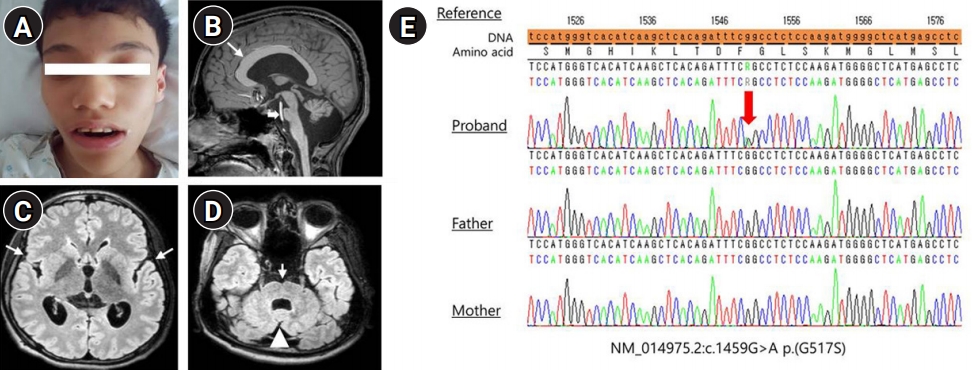
A 19-year-old man was the first of three children of healthy non-consanguineous parents. The patient was born via vaginal birth at 40 weeksŌĆÖ gestational age, with a birth weight of 2.8 kg. Although his fetal movements were reduced, perinatal problems were not detected. He showed poor feeding and hypotonia with upward gaze during the neonatal period. He started to control his head at 1 year of age and sit without support when he was 3 years old. He experienced his first generalized tonic clonic seizure at the age of 8 years, which recurred after 1 year. His mother recollected that magnetic resonance imaging (MRI) performed at that time revealed ventriculomegaly, and electroencephalography (EEG) showed slow background rhythms. He was administered topiramate, an antiseizure drug, for 2 years but had been weaned off the medication. At the age of 17 years, he was referred to our hospital for the evaluation of the recurrence of two febrile generalized tonic clonic seizures lasting for 2 minutes. His development progressed slowly, and he could stand up with support. However, he exhibited language and intellectual impairments and his speech consisted only of babbling. He had spastic diplegia with muscular atrophy in the lower extremities. He also exhibited microcephaly (52 cm, <the third percentile), low weight (32 kg, <the third percentile), and short stature (152 cm, <the 10th percentile). He also had facial dysmorphism (Fig. 1A), dolichocephaly, scoliosis, oculomotor apraxia, strabismus, and small testes without secondary sexual characteristics. MRI revealed a mega-corpus callosum, focal cortical dysplasia, and cerebellar hypoplasia (Fig. 1B-D). Slow and disorganized background rhythms with parietal vertex sharp waves were observed on EEG. Laboratory investigations revealed vitamin D deficiency (25-hydroxy vitamin D3, 6.44 ng/mL; reference range, 30 to 150 ng/mL), abnormal gonadotropin-releasing hormone stimulation (peak simulated luteinizing hormone, 16.5 mIU/mL; reference range, 3.3 to 5 mIU/mL) and testosterone levels (0.039 ng/mL; reference range, 2.49 to 8.36 ng/mL), normal insulin-like growth factor (IGF)-1, and low IGF-binding protein-3 (2,860 ng/mL; range, 3,200 to 8,700 ng/mL). His karyotype was 46,XY, but microarray testing was not performed due to parental refusal. WES showed de novo heterozygous variants in MAST1 (NM_014975.2:c.1549G>A). Sanger sequencing of these variants obtained from the peripheral blood of patient and his parents revealed a de novo mutation in the patient, while both parents had the wild-type gene (Fig. 1E). He received valproic acid (750 mg daily) for recurring seizures and was seizure-free for 18 months. He also received vitamin D owing to deficiency detected on laboratory testing and testosterone hormonal therapy for delayed puberty.
MCC-CM due to MAST 1 mutation is characterized by delayed development, intellectual disability, impaired language abilities, and specific brain abnormalities (Table 1) [2-4]. Patients with MAST1 mutations can present with varying phenotypes, including MCC-CM, microcephaly and cerebellar hypoplasia, autism spectrum disorder, global developmental delay, and cerebral palsy [2]. Our patient exhibited MCC-CM and microcephaly, which were diagnosed by the detection of heterozygous variants in MAST1 (c.1549G>A). MAST1 is expressed in multiple organs, including the spleen, kidney, testis, and skeletal muscle [2]. Our patient exhibited small testes and lack of secondary sexual characteristics, which could be due to MAST1 mutations in the testes.
MAST 1, located on chromosome 19p13.13, encodes a microtubule-associated serine/threonine kinase protein expressed in the postmitotic neurons in the developing brain. This protein consists of an N-terminal domain of the unknown function (DUF1908), a serine/threonine kinase domain and a post-synaptic density protein-95/discs large/zona occludens-1 (PDZ) domain. It supports a PDZ-dependent interaction with other proteins and also interacts with tumor suppressor phosphatase and tension homolog (PTEN) via the PDZ domain to facilitate the functioning of the phosphorylation of the interacting proteins [2,5]. We suggest that the reduction in PTEN function, which encodes a negative regulator of the mechanistic target of rapamycin signaling pathway, may influence cortical malformation and social-behavioral problems because a significant reduction was observed in the levels of the MAST family of proteins in MAST1 mutant rodents compared to their wild-type counterparts [2]. Further research is needed to determine the genetic function and mechanisms affecting the disease phenotypes.
In conclusion, we report the first case of a Korean patient with MAST1 gene mutation leading to MCC-CM without secondary sexual characteristics.