Introduction
Movement disorders often arise from the basal ganglia nuclei or when their connections malfunction, resulting in hypokinetic, hyperkinetic, or dystonic disorders [1]. A seizure is the result of abnormally excessive or synchronous neuronal activity in the brain, defined as ŌĆ£momentary arising of signs and/or symptoms,ŌĆØ whereas epilepsy is characterized by one or more seizures with a relatively high recurrence risk [2]. Although they are caused by a number of different conditions, both movement disorders and seizures present abnormal movements with coinciding phenomenology [3]. Both movement disorders and epilepsy occur in many genetic disorders ranging from inborn errors of metabolism to developmental and epileptic encephalopathies, epilepsy syndrome with stereotypies, and paroxysmal dyskinesias [3,4].
Paroxysmal dyskinesias are an important disease paradigm associated with overlapping movement disorders and seizures [5]. In many paroxysmal dyskinesias, movement disorders and seizures occur simultaneously at the time of, before, or even a long time after, diagnosis. The most well-known is paroxysmal kinesigenic dyskinesia (PKD) in which PRRT2 mutations have been identified. In this case, infantile convulsions often occur in advance [6]. With the development of exome sequencing in recent years, many genetic abnormalities identified in epilepsy that occur in conjugation with paroxysmal dyskinesias have been identified [3,7-9]. This review aims to gather the most updated literature regarding paroxysmal movement disorders, and seizures and we will discuss the genetic abnormalities that come with seizures for each paroxysmal movement disorders. Table 1 summarizes the clinical manifestations of each epilepsy-paroxysmal movement disorders.
Clinical overview
Paroxysmal dyskinesias have distinct features with episodic occurrences of involuntary extrapyramidal movements [3]. It is a heterogeneous disorder group characterized by episodes of abnormal involuntary movements, such as chorea, dystonia, and ballism [3]. Most of these movements do not cause loss of consciousness but sometimes a sensory aura precedes them [3]. Some clinicians may mistake these movements for focal seizures, either focal aware seizure or focal impaired awareness seizure [3]. In such cases, the absence of ictal discharges in a scalp electroencephalography may be helpful in the diagnosis of paroxysmal dyskinesias [2].
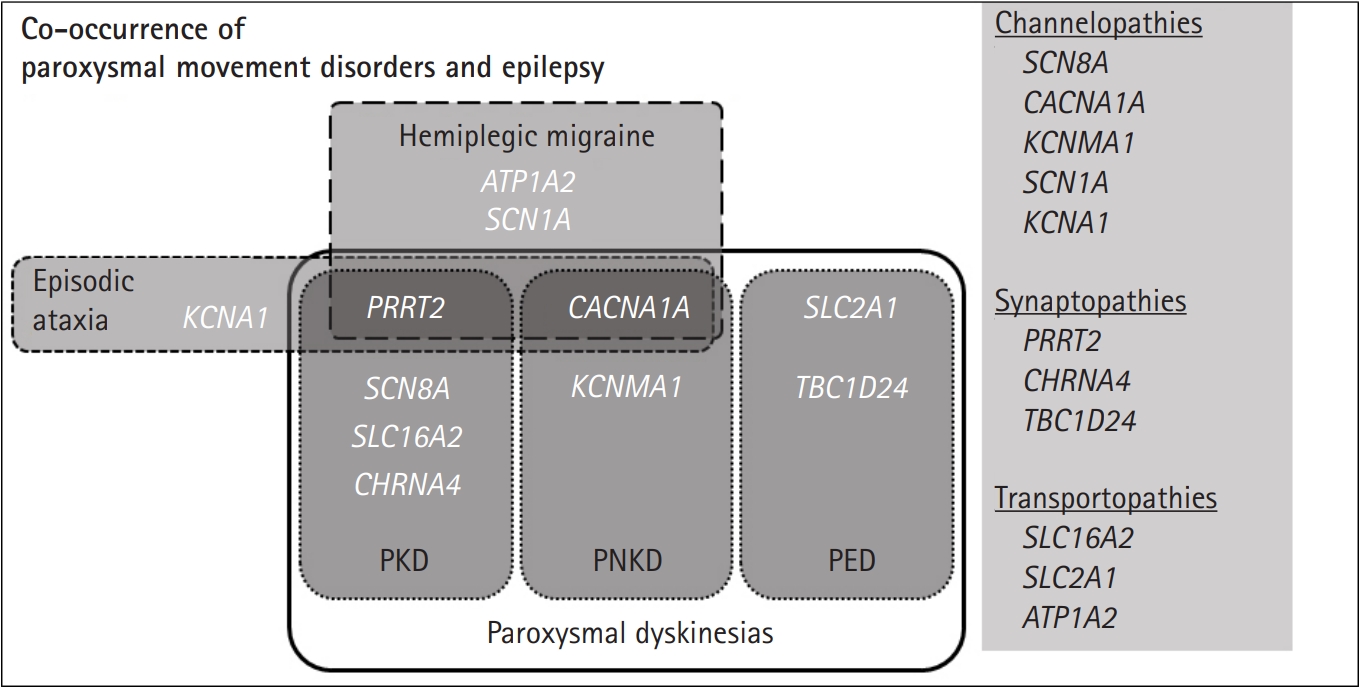
Paroxysmal dyskinesias are divided into three clinical syndromes (Fig. 1): PKD, paroxysmal non-kinesigenic dyskinesia (PNKD), and paroxysmal exercise-induced dyskinesia (PED) [6,8]. PKD is the most common type and is caused by voluntary movements, such as standing from a seated position or transitioning from walking to running. PKD attacks usually develop during childhood and are well controlled by carbamazepine [5,10]. Infantile convulsions, often with choreoathetosis, frequently precede PKD [6]. PNKD attacks are usually triggered by alcohol, coffee, or strong emotions [6], and can often last longer than PKD attacks ranging from 10 minutes to an hour and even up to 12 hours [6]. However, their frequency is low, typically occurring only a few times a year [6]. Of the three paroxysmal movement disorders, PED is the rarest. PED attacks are induced by physical exertion after long periods of exercise with migraines, hemiplegia, ataxia, and epilepsy being associated with PED [6,11].
In terms of co-occurrence with epilepsy, patients with mutations in PRRT2 [9,12], SCN8A [13,14], SLC16A2 [15,16], and CHRNA4 [17] have been reported in PKD (Fig. 1). In PNKD, CACNA1A [18] and KCNMA1 [19]-related diseases have been reported, while SLC2A1-related glucose transporter-1 (GLUT1) protein deficiency [6] and recently, TBC1D24 mutations, have been reported in PED [20]. Other paroxysmal movement disorders associated with epilepsy include hemiplegic migraine and episodic ataxias. A combination of familial hemiplegic migraine and epilepsy have been found in PRRT2, CACNA1A, SCN1A, and ATP1A2 mutation-positive patients [8,21]. In addition, PRRT2 [8], CACNA1A [18], and KCNA1 [8,22,23] mutations are mainly responsible for the co-occurrence of episodic ataxias and epileptic seizures [24,25].
Historically, paroxysmal dyskinesias and other episodic neurological disorders have been considered an ion channel dysfunction [5]. Accordingly, it has been suggested that the relationship between paroxysmal dyskinesias and epilepsy is in the form of ŌĆ£basal ganglia epilepsy,ŌĆØ meaning that paroxysmal dyskinesias attacks are due to the altered functions of ion channels in both the cortex and subcortex [26]. However, with the discovery of the major paroxysmal dyskinesias genes PRRT2 and SLC2A1, the hypothesis of channelopathy lost its strength because neither encoded ion channels [5]. While novel mutations in genes related to ion channel function, such as KCNA1, KCNMA1, SCN8A, and CACNA1A, have recently been described, genes encoding synaptic proteins/receptors, such as PRRT2, CHRNA4 and transporters including, SLC2A1, SLC16A2, and ATP1A2, have also been identified (Fig. 1) [5]. As the understanding of the genetic basis of epilepsy syndrome and paroxysmal dyskinesias increase, it provides insights into the shared mechanisms behind the two conditions and reveals the role of ion channels and the proteins associated with vesical synapse or energy metabolism [5,6,14,27].
PKD and epilepsy
1. PRRT2
PRRT2, which represents proline-rich transmembrane protein 2, encodes a transmembrane protein involved in synaptic transmission, although its function is relatively unknown [12]. Genetic mutations of PRRT2 not only occur in PKD patients, but also in most cases of benign infantile familial seizure, infantile convulsions with paroxysmal choreoathetosis (infantile convulsion with choreoathetosis), frequent cases of hemiplegic migraine, and in a minority of cases of episodic ataxias, childhood absence epilepsy, paroxysmal torticollis, and febrile seizure [5,8,12].
PKD affects about 1:150,000 in the general population [12]. As is well known, the major gene responsible for PKD is PRRT2, and according to the case ascertainment, the frequency ranges from about 40% to 90% or more [5,6,12,28]. A majority of PRRT2 cases have an obvious kinesigenic trigger with anxiety or prolonged exercise triggering PKD attacks in up to 40% of cases [5]. PKD attacks are typically very short (i.e., less than 1 minute), but with a high frequency (i.e., occurs more than once daily). They usually consist of chorea and dystonia but also rarely athetosis, ballism, hemiballism, tongue movements, perioral dyskinesias, and clawing of the hands or a frozen gaze [5,12]. The attacks are bilateral or sometimes unilateral and tend to generalize [5,12]. Symptoms most commonly manifest shortly before or during puberty in PRRT2 mutation carriers with less than 5% of PRRT2-associated cases experiencing an onset after 18 years of age [12]. Patients often experience several attacks a day but regardless of treatment, the frequency decreases with advancing age after puberty [5].
Benign infantile familial seizure is inherited in an autosomal dominant pattern with up to 80% of cases exhibiting mutations in PRRT2 [3,12]. It is characterized by seizures that occur between 3 and 12 months of age [10] and involve brief, focal motor manifestations accompanied by cyanosis, hypertonia, and limb jerks [3,12]. They can occur in clusters of multiple seizures, up to eight to ten seizures per day, which occur every 2 to 3 hours on average [10]. However, patients have an excellent response to antiepileptic drugs and seizures generally resolve by the age of 2 years [12,29]. That aside, patients demonstrate normal developmental outcomes, neurological examinations, brain imaging patterns, and electroencephalography background signals [10].
Similarly, nearly 90% of infantile convulsion with choreoathetosis syndrome patients have PRRT2 mutations [5]. Overall, the clinical findings and genetic features overlap with cases of PKD and benign infantile familial seizure. This syndrome is characterized by the development of PKD after infantile convulsions (PKD usually develops by the age of 5 years) as some epileptic seizures may exhibit at a much later age than typical benign infantile familial seizure [5]. Remission rate for treated infantile convulsion with choreoathetosis cases is up to 89%, which means that only a small number of patients maintain partial response to therapy [12].
The PRRT2 gene has recently been implicated in the shared pathophysiology of epilepsy and hemiplegic migraine [30]. Although PRRT2 mutations are rare, they have also been identified in hemiplegic migraine with PKD, and/or benign infantile familial seizure [31]. Interestingly, CACNA1A, ATP1A2, or SCN1A genes, or in certain combinations, have been found in approximately 75% of familial hemiplegic migraine patients and also in a smaller number of sporadic hemiplegic migraine patients [32].
Historically, PKD, infantile convulsion with choreoathetosis, and benign infantile familial seizure have been considered to be allelic disorders because they occurred together in some families and were linked to the same region on chromosome 16p11.2-q12.1 [33,34]. In 2011, Chen et al. [9] first identified mutations of PRRT2 located on chromosome 16p11.2 in eight Chinese PKD families by whole exome sequencing. A significant number of PRRT2 mutations associated with loss-of-function and missense amino-acid change mutations have since been identified [6]. To date, a total of 97 different PRRT2 mutations have been reported [35], of which c.649dupC is a hotspot. It is found in 60% to 80% of PRRT2-associated PKD, benign infantile familial seizure, and infantile convulsion with choreoathetosis patients [12]. In addition, deletion mutation at the same location (c.649delC; approximately 4%) and at the more proximal part (c.291delC; approximately 2%) have also been identified [12]. However, a c.579dupA mutation of the PRRT2 gene presents more commonly in infantile convulsion with choreoathetosis [12].
The PRRT2 gene comprises of four exons that encodes the 340-amino acid, proline-rich transmembrane protein 2 [12]. PRRT2 is found throughout the central nervous system, especially at high expression levels in the cortical layers of the cerebral cortex, basal ganglia, and cerebellum [9]. The contribution of alteration of the basal ganglia-thalamocortical circuit to the pathogenesis of PRRT2-associated PKD has been proposed [36,37]. It is mainly recognized in the axons but not in the dendrites of neurons at the subcellular level [12]. A yeast two-hybrid assay found that PRRT2 interacts with the synaptic t-SNARE protein synaptosomal-Associated Protein, 25kDa (SNAP25) [38,39]. SNAP25 alters neurotransmitter release and calcium channel dynamics, which affects vesicle synapse accordingly [40]. It has been suggested that PRRT2 mutations impair SNAP25 function by altering Cav2.1 activity, which lead to neuronal hyperexcitability and subsequently cause epilepsy, PKD, or other paroxysmal movement disorders [41]. In addition to the relation between PRRT2 and SNAP25, many studies proposed that PRRT2 has postsynaptic roles in alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor signaling [42,43].
In addition to the roles at the synapse, recent studies have suggested that PRRT2 interacts with ion channels, such as Nav1.2/Nav1.6 channels, major regulators of the excitability of excitatory neurons, but not with Nav1.1 channels, which is essential for the excitability of inhibitory neurons [44]. Thus, the disturbance in cellular excitability by lack of negative modulation of Na+ channel was assumed to be the main pathogenetic mechanism of paroxysmal character [44].
Currently, there is no clear evidence to propose genotype-phenotype correlations [12,35]. Variable phenotypes are found within and between families with the same PRRT2 mutations [10]. Patients with a 16p11.2 deletion not only have PRRT2-PKD but also additional clinical features, including developmental delay, intellectual disability, and/or autism spectrum disorder [45,46]. Analogously, patients with biallelic PRRT2 mutations often show more severe phenotypes, including intellectual disability, episodic ataxia, and different seizure types [47,48].
2. SCN8A
Pathogenic variants of SCN8A, which encode the sodium channel voltage-gated ╬▒8-subunit (Nav1.6), have originally been described in patients with developmental and epileptic encephalopathies [13,49]. However, recent studies showed that SCN8A pathogenic variants have a wide phenotypic spectrum ranging from developmental and epileptic encephalopathies to behavioral disorders or movement disorders [50]. A variety of seizure types along with episodes of paroxysmal dystonia, sometimes resembling PKD, can be caused by mutations in SCN8A [5,14].
Clinical syndromes of PRRT2 mutations (PKD, benign infantile familial seizure, and infantile convulsion with choreoathetosis) have been reported in patients with SCN8A mutations [14]. Some individuals manifest either with afebrile focal or generalized tonic-clonic seizures during the first 2 years of life, often having paroxysmal dyskinetic/dystonic episodes in puberty, which can be triggered by movement initiation (e.g., stretching) and/or emotional stimuli [14]. Choreo-dystonia and dystonic dyskinesias present in some individuals with SCN8A-related epileptic encephalopathies [13], which suggests that episodic movement disorders also occur with pathogenic variants of SCN8A.
Patients with mutations in PRRT2 and SCN8A have similar clinical manifestations, but there are many significant findings that help differentiate the diagnosis [13]. SCN8A patients have a variety of seizure types along with focal, tonic, clonic, myoclonic, and absence seizures, and they are usually refractory to antiepileptic drugs [14]. Furthermore, regardless of whether early development was normal, patients with SCN8A mutations develop moderate to severe intellectual disability [13] and might coincide with non-epileptic paroxysmal movement disorders, including dystonia and ataxia [13]. In addition, most SCN8A mutations are de novo but only one case of somatic mosaicism in an unaffected parent has been reported [13]. All these characteristics make SCN8A-related disorders different to that of PRRT2.
3. SLC16A2
The SLC16A2 gene located on chromosome Xq13.2 encodes for monocarboxylate transporter 8 (MCT8), which is an active transporter protein in humans [16]. MCT8 transports diverse iodo-thyronines including triiodothyronine (T3) and thyroxine (T4) [16]. MCT8 deficiency, also known as Allan-Herndon-Dudley syndrome, is characterized by an increased level of free T3, normal to low free T4, low reverse T4, and normal to elevated thyroid stimulating hormone without any signs or symptoms of congenital hypothyroidism [15]. Clinical characteristics consist of variable mental retardation, hypotonia, spasticity and pyramidal signs, facial/neck weakness, ataxia, and paroxysmal purposeless movements with a static or slow progressive course [15,16].
Epileptic seizures, including febrile seizure, myoclonic seizure, and generalized tonic-clonic seizures, are found in almost half of Allan-Herndon-Dudley syndrome [16,51,52]. Involuntary movements, including dystonic and/or athetoid and characteristic paroxysms or kinesigenic dyskinesias, are common in affected males [15,53]. Attacks manifest through stretching of the body, opening of the mouth, or extending or flexing of the limbs for 1 to 2 minutes [53]. Somatosensory stimuli, including the changing of clothes or diapers, or lifting the affected child can trigger attacks [53].
4. CHRNA4
CHRNA4, which encodes the ╬▒4 subunit of the neuronal nicotinic acetylcholine receptor (nAChR), frequently assembles with the ╬▓2 subunit (encoded by CHRNB2) to form a heteropentamer ╬▒4 ╬▓2-nAChR [56]. Pathogenic variants of CHRNA4 have been found to be the major cause of autosomal dominant nocturnal frontal lobe epilepsy, which causes frequent motor seizures during non-rapid eye movement sleep [57].
Recently, Jiang et al. [17] found that co-occurrence of genetic epilepsy with febrile seizures plus and PKD have CHRNA4 mutations in PRRT2-negative families. Different seizure types were observed, including recurrent febrile seizures, which occurred between the ages of 3 and 7 years, afebrile seizures, including myoclonic seizures, which occurred between the ages of 6 and 11 years, and generalized tonic-clonic seizures, which occurred after the age of 14 years [17]. Symptoms of choreoathetosis and dystonia were mostly triggered by sudden movements and only occurred during daytime [17]. The attacks usually lasted for less than 30 seconds and did not cause any loss of consciousness and with oxcarbazepine monotherapy, they were markedly controlled [17].
PNKD and epilepsy
1. KCNMA1
The KCNMA1 gene encodes the ╬▒ subunit of the large conductance, voltage, and calcium-sensitive potassium channel, which is also activated by the concentration of cytosolic Mg2+, and is known to be predominantly expressed in the amygdala, caudate nucleus, cerebral cortex, hippocampus, hypothalamus, spinal cord, and Purkinje cells in the cerebellum [58,59]. Heterozygous mutations in KCNMA1 were first reported in a large family with generalized epilepsy and PNKD [27]. Recently, homozygous KCNMA1 mutations was illustrated in patients with cerebellar atrophy, developmental delay, and seizures [60].
Mutations in the KCNMA1 gene produces a syndrome of PNKD and epilepsy, either in the form of absence seizures or generalized tonic-clonic seizures [5,19]. In 2005, Du et al. [27] reported a family with an autosomal dominant form of generalized epilepsy and paroxysmal dyskinesias carrying a mutation in the KCNMA1 gene. Clinically, paroxysmal dyskinesias attacks that involve KCNMA1 mutations were described to resemble the non-kinesigenic variant with alcohol being a possible trigger [27]. Recent studies established a correlation of the homozygous KCNMA1 mutation with cerebellar ataxia, cortico-cerebellar tract atrophy, developmental delay, paroxysmal dyskinesia, and variable epilepsies, including absence, myoclonic, atonic, tonic, and generalized tonic-clonic seizures [19,60]. In addition, both gain- and loss-of-function have been proposed as the underlying molecular mechanism behind the channelopathy, which causes an increase in excitability [60].
PED and epilepsy
1. SLC2A1
GLUT1 deficiency syndrome is caused by mutations in SLC2A1, which presents early-onset refractory seizures, PED, and movement disorders [6]. SLC stands for solute carrier, while 2A1 represents the family number 2 and member number 1 in the family. As GLUT1 is one of the proteins located on the blood-brain barrier, GLUT1 deficiency syndrome was previously described in association with infantile epilepsy with low cerebrospinal fluid glucose [61]. Both epilepsies, particularly early-onset absence, and PED co-occur in families and individuals [5].
This disorder is usually classified into two groups: classical and nonclassical. Classical or typical GLUT1 deficiency involves infantile-onset, pharmaco-resistant epilepsy, intellectual disability, microcephaly, and complex movement disorders; while nonclassical or atypical GLUT1 deficiency involves paroxysmal movement disorders, atypical childhood absence epilepsy, and myoclonic astatic epilepsy [61]. Infantile-onset epilepsy can be alleviated during childhood but movement disorders tend to emerge later, which may be due to changes in brain metabolism over time [62].
Approximately 90% of patients have clinical seizures, mainly generalized tonic-clonic seizures followed by absence, myoclonic, and focal onset seizures [61]. The PED attacks usually consist of choreoathetosis and dystonia, which mainly affect the lower limbs, and are typically triggered by sustained exercise [5]. Notably, this disturbance might be misdiagnosed as epileptic myoclonic seizures [63]. The combination of epilepsy with a possible family history and PED in the setting of an unremarkable neurological examination, along with low cerebrospinal fluid glucose concentration, represents an important clinical clue to raising the correct diagnostic suspicion [5]. An early diagnosis of GLUT1 deficiency is crucial given that the syndrome can be well managed with a ketogenic diet [5,61].
Although isolated PED caused by SLC2A1 mutations are rare, episodes of PED in those suffering from GLUT1 deficiency syndrome are common but often go unnoticed in the setting of epilepsy or more severe findings [61]. Isolated dystonia after exercise that usually only affects the lower limbs have also been observed in carriers of SLC2A1 mutations that cause early-onset Parkinsonism or dopa-responsive dystonia; however, these are rather unusual initial presentations of these conditions [8].
No clear-cut phenotype-genotype correlations have been established [64]. Patients exhibited interindividual phenotypic variability despite having the same mutations, which suggests the presence of genetic modifiers, such as secondary genes [61,64]. Therefore, the genotype does not always predict the phenotype [61].
2. TBC1D24
The gene TBC1D24 is involved in the regulation of synaptic vesicle trafficking by interacting with GTPase in brain and somatic development [65,66]. Genetic mutations in TBC1D24 have been associated with multiple phenotypes with epilepsy as the main clinical manifestation [20,66]. Epilepsy aside, TBC1D24 is also associated with deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures (DOORS syndrome), as well as nonsyndromic deafness [65].
The types of seizures and epilepsies are diverse. Seizure types include infantile spasms, febrile convulsions, myoclonic, clonic, tonic, absence, tonic-clonic seizures with or without apparent focal onset, and focal seizures with retained or impaired awareness [65]. Myoclonic or clonic seizures are the most frequent seizure types, which include infantile and progressive myoclonic epilepsies, as well as familial epilepsy of infancy with migrating focal seizures and are often unresponsive to medication [65].
TBC1D24 epilepsy syndromes occur with both compound heterozygous and homozygous recessive mutations. More than 50 missense and loss-of-function mutations have been described and associated with the exercise-induced dystonia phenotype, which persist into adulthood according to a long clinical follow-up study [20]. The additional diversity of TBC1D24 phenotypes might be due to its broader expression patterns; TBC1D24 is expressed in several human tissues with the highest expression occurring in the brain in multiple cerebral areas, including all layers of the cerebral cortex and the hippocampus [66]. In a Drosophila model, some mutations of TBC1D24 cause activity-induced locomotion and synaptic vesicle trafficking defects, which is consistent with exacerbated oxidative stress sensitivity, suggesting that these mutations cause dysfunctional, sustained movement disorders [20].
Other paroxysmal movement disorders and epilepsy
1. Familial hemiplegic migraine and epilepsy
CACNA1A, ATP1A2, SCN1A, and PRRT2 genes often contain one or more mutations in both epilepsy and hemiplegic migraine patients [8,21,41]. These shared mutations identified in epilepsy and migraine cases suggest that there is a common genetic basis for these conditions. There are two categories of migraine: migraine with and without aura, and hemiplegic migraine is a rare form of migraine with aura [41].
1) ATP1A2
The ATP1A2 gene is located on chromosome 1q23 and encodes for the ╬▒2 subunit of Na+/K+ ATPase, which consists of an ╬▒ and a ╬▓ subunit. ATP1A2 mutations were identified in families with familial hemiplegic migraine, called familial hemiplegic migraine type 2 [67,68].
The incidence of epilepsy is increased in families with familial hemiplegic migraine type 2, where approximately 20% experience seizures, such as focal seizures, benign infantile familial seizure, and high fever convulsions [68]. In a family with familial hemiplegic migraine type 2, one member had focal epilepsy as a child and electroencephalography revealed a focal migratory epilepsy-like discharge waveform [68].
Since maintaining the correct concentrations of Na+ and K+ via the Na+/K+ ATPase system is crucial for the ability of astrocytes to clear extracellular glutamic acid, an abnormal Na+/K+ ATPase system function disrupts the K+ gradient and impairs glutamate clearance, which likely contributes to the development of familial hemiplegic migraine and epilepsy [41].
2) SCN1A
SCN1A, which encodes the ╬▒1 subunit of the sodium channel, is associated with a range of human diseases [69]. The most well-recognized epilepsy phenotype associated with SCN1A is the Dravet syndrome but it also results in several other epilepsy syndromes ranging from self-limited and pharmacoresponsive epilepsies, such as genetic epilepsy with febrile seizures plus, Dravet syndrome, myoclonic-atonic epilepsy, and epilepsy of infancy with migrating focal seizures. SCN1A disorders also result in other neurological disorders such as hemiplegic migraine, intellectual disability, and autism spectrum disorder [69].
Familial hemiplegic migraine type 3 is caused by heterozygous pathogenic variants of the SCN1A [70]. In contrast to familial hemiplegic migraine due to CACNA1A and ATP1A2 mutations, in the few patients with familial hemiplegic migraine type 3 carrying SCN1A mutations and presenting with seizures [69], hemiplegic migraine attacks are always independent from seizures and the two phenotypes do not generally overlap temporally [69].
All SCN1A mutations reported in familial hemiplegic migraine type 3 are missense mutations. Most experimental results show that they cause a gain-of-function of Nav1.1 [71]. Cellular and animal data point to an increased excitability of gamma-aminobutyric acid (GABAergic) neurons in familial hemiplegic migraine type 3, which is a different mechanism from that seen in epileptogenic Nav1.1 mutations [69].
Episodic ataxia and epilepsy
Episodic ataxia is a rare neurological condition characterized by recurrent spells of truncal ataxia and incoordination. PRRT2, CACNA1A, and KCNA1 mutations are mainly responsible for co-occurrence of episodic ataxias and epileptic seizures [24,25].
1) CACNA1A
CACNA1A is located on chromosome 19p13 and encodes for the ╬▒1 subunit of the Cav2.1 P/Q-type voltage-gated calcium channel [18]. Pathogenic variants of CACNA1A are associated with three allelic autosomal dominant conditions: episodic ataxias type 2, spinocerebellar ataxia type 6, and familial hemiplegic migraine type 1 [18]. Other paroxysmal disorders, including benign paroxysmal torticollis of childhood, benign paroxysmal tonic upward gaze, and epilepsy, are also associated with CACNA1A mutations [8].
Patients with episodic ataxia type 2 usually experience intermittent episodes of ataxia and nystagmus that can last from minutes to days during childhood or early adulthood [8,72]. It usually develops with dysarthria, tinnitus, dystonia, hemiplegia, and headache with the frequency of attacks varying from once or twice a year to three or four times a week [73]. Typically, exertion, stress, heat, fever, alcohol, caffeine, or drugs, such as phenytoin, can trigger these episodes [73]. Myokymia (fine twitching or rippling of muscle) is absent on physical examination or electromyographic studies [59]. Episodic ataxia type 2 attacks can be interrupted or reduced in frequency and severity by acetazolamide or 4-aminopyridine administration [73]. Few patients have epileptic encephalopathy with generalized absence or focal seizures with or without generalized tonic-clonic seizures and/or intellectual disabilities [18,74].
About half of families with familial hemiplegic migraine have heterozygous pathogenic missense variants in CACNA1A, which are called familial hemiplegic migraine type 1 [73]. Episodic hemiplegia occurs with one or more sensory auras such as hemianopsia, hemisensory deficit, or aphasia in familial hemiplegic migraine type 1 [73].
Benign paroxysmal torticollis of childhood is a rare paroxysmal disorder characterized by recurrent episodes of head tilt accompanied by general symptoms that remit spontaneously [75]. The rare association with gain-of-function and loss-of-function CACNA1A mutations has been reported [75].
Benign paroxysmal tonic upward gaze was initially described as a benign phenomenon with negative investigations and eventual complete resolution of symptoms [76]. Later publications demonstrated that a similar clinical feature may arise from structural brain lesions, channelopathies, neurotransmitter disorders, and epileptic seizures [76]. CACNA1A mutations were detected in infants and young children with benign paroxysmal tonic upward gaze especially if associated with developmental delay, cerebellar signs, and other types of paroxysmal events [76].
Patients with CACNA1A mutations experience a high rate of different seizure types, involving febrile seizures, epileptic encephalopathy, generalized absence seizure, and focal seizures with or without generalized tonic-clonic seizures [18].
Pathological manifestations are explained by the synaptic dysfunction caused by the loss of Cav2.1 channels in particular cell types [77-79]. In conditional mutant mice, selective deletions of CACNA1A in cerebellar granule cells or Purkinje cells reduce the excitatory drive and neurotransmitter release, which cause ataxia and dyskinesia [80,81]. While GABA release is impaired, generalized epilepsy in cortical and hippocampal GABAergic interneurons [77].
2) KCNA1
Episodic ataxia type 1, which is also called ataxia with myokymia, is caused by heterozygous pathogenic variants in KCNA1, which encodes a potassium channel [82]. It is characterized by brief attacks (<15 minutes) of ataxia and dysarthria that can occur up to 15 times per day [82]. Attacks can occur spontaneously or be triggered by anxiety, exercise, startle, and/or intercurrent illness [82]. Onset typically occurs in late childhood and early adolescence, and symptoms usually remit during the second decade [83]. Between attacks, widespread myokymia of the face, hands, arms, and legs occur. Electromyographic studies reveal myokymia, so called neuromyotonia [82]. Phenytoin can control symptoms; acetazolamide is also effective [84].
Episodic ataxia type 1 may be associated with epilepsy as tonic-clonic and focal seizures, one isolated episode of photosensitive epilepsy [85], as well as symptoms, such as head-turning, eyes deviating to the same side, flickering eyelids, lip-smacking, apnea, and cyanosis, have been reported [86]. Prolonged episodes of more than 30 minutes have been reported in individuals with severe early-onset epilepsy, albeit without the typical ataxia [87].
The molecular mechanisms of episodic ataxia type 1 are described as impaired channel function and reduced outward K+ flux through the channel [85,88]. In a mouse model of episodic ataxia type 1, altered motor performance and impaired cerebellar GABAergic transmission from the basket cells to the Purkinje cells was found [89], resulting in spontaneous myokymic activity, which was exacerbated by fatigue, ischemia, and low temperature [90]. However, although a similar phenomenon to the spread of acidification in the cerebellar cortex has been described, the causes of triggering the paroxysms of ataxia remain unknown [91].
Conclusion
Several genetic disorders were identified as co-occurrences of epilepsy and paroxysmal dyskinesias. Disease and associated genes are as follows: (1) PKD: PRRT2, SCN8A, SLC16A2, CHRNA4; (2) PNKD: CACNA1A, KCNMA1; (3) PED: SLC2A1, TBC1D24; (4) hemiplegic migraine: PRRT2, CACNA1A, SCN1A, ATP1A2; and (5) episodic ataxia: PRRT2, CACNA1A, KCNA1. These conditions are divided into three pathomechanisms: (1) channelopathy: SCN8A, CACNA1A, KCNMA1, SCN1A, KCNA1; (2) synaptopathy: PRRT2, CHRNA4, TBC1D24; and (3) transportopathy: SLC16A2, SLC2A1, ATP1A2.