Dihydropteridine reductase (DHPR) deficiency is a severe form of hyperphenylalaninemia due to impaired renewal of a substance known as tetrahydrobiopterin (BH4), caused by mutations in the quinoid dihydropteridine reductase (QDPR) gene [1]. If little or no BH4 is available to facilitate processing phenylalanine (Phe), this amino acid can accumulate in the blood and other tissues, and the levels of neurotransmitters (dopamine, serotonin) and the folate in cerebrospinal fluid also decrease [1]. Symptoms such as mental retardation, with motor impairment and epilepsy occur in untreated patients with severe forms of DHPR deficiencies [2].
Thus, many experts misunderstand if seizure or movement disorders occur in DHPR deficiency patients, it may be because of uncontrolled disease and miss other pathologic conditions that may accompany it. Although DHPR deficiency can cause neurological problems, an identification of other causes is required.
Here, we report the case of an adolescence patient impacted by DHPR deficiency, who had experienced absence seizures and myoclonic seizures, and early suspected dopamine deficiency tremor. But after identifying ictal epileptiform discharges on the electroencephalography (EEG), it was treated as idiopathic generalized epilepsy.
A 15-year-old man was admitted to Soonchunhyang University Seoul Hospital. At the age of 3 months, he experienced the first seizure, characterized by brief psychomotor arrest and right hand tremor. He had no family history of seizures or genetic disorders. Brain magnetic resonance imaging (MRI) and EEG showed normal results. The seizures were not controlled by antiepileptic drugs, including topiramate, valproate, and lamotrigine; therefore, he discontinued the medication. A year later, he was diagnosed with hyperphenylalaninemia in other pediatric unit. His blood Phe level on admission was 12.9 mg/dL (normal <2 mg/dL). BH4 loading test was negative at that time, so he was managed with just a low Phe diet. However, at the age of 7 years, he was diagnosed with DHPR deficiency in our hospital. His basal blood Phe level was 10.3 mg/dL, and 8 hours after BH4 loading, the Phe level had dropped to 1.9 mg/dL. This represented a 72% reduction in Phe level, which is consistent with BH4 deficiency findings. Further tests showed normal neopterin with elevated biopterin levels. The sanger sequencing of the patient identified two heterozygous mutations (c.253T>C and c.515C>T) on the two alleles of QDPR gene, which were both classified as likely pathogenic. Treatment started promptly and included levodopa (2 mg/kg, four times daily), 5-hydroxytrptophan (2 mg/kg, four times daily), folate (10 mg/day), and BH4 supplementation (20 mg/kg, twice daily), along with a low Phe diet. BH4 dosage was adjusted to keep his serum Phe concentration less than 2 mg/dL, with a dietary restriction of Phe. With this therapy, seizure-like movements had already significantly improved, and blood Phe levels had dropped significantly to 2 mg/dL. Since then, the frequency had decreased from 10 times a day to once or twice monthly, but abnormal movements were observed, such as staring blankly or trembling in both hands. As the EEG was normal at that time and the serum prolactin level showed a rise, the levodopa dose was increased with a good response. Despite delay in the treatment, he showed no severe mental impairment.
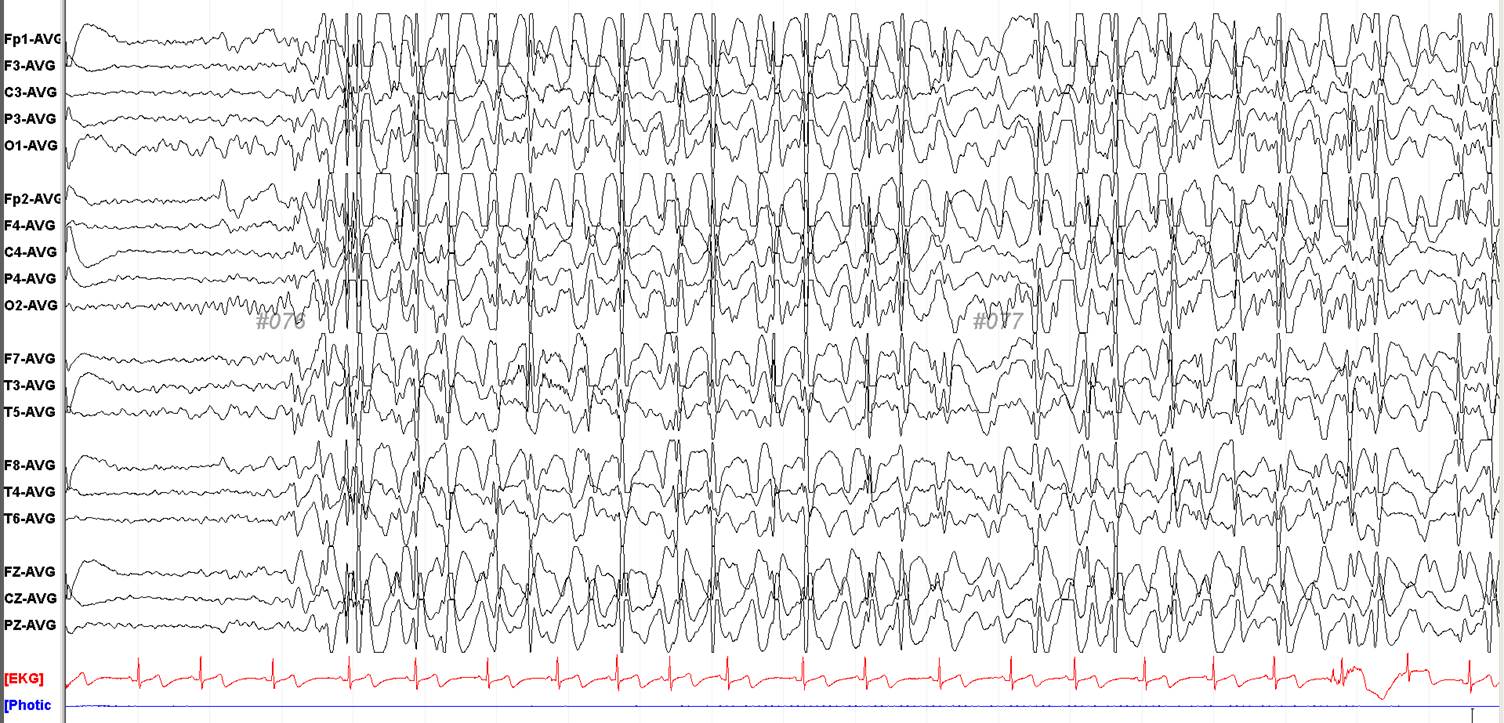
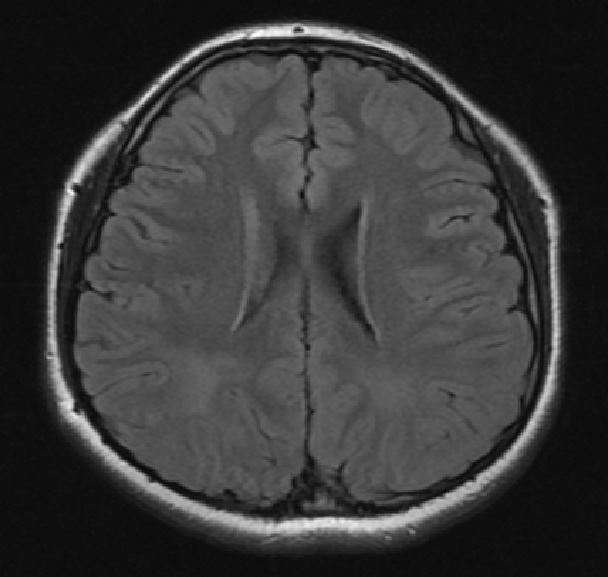
However, the seizures became frequent, and he experienced several daily episodes of staring blankly for a moment, flinching both legs, and trembling in both hands by the age of 15 years. At that time, he was taking levodopa (40 mg/day), 5-hydroxytroptophan (40 mg/day), and folate (10 mg/day). His Phe level was 4.9 mg/dL and prolactin level was 35.27 ng/mL (normal 2 to 18). Given the assumption that seizures may not be controlled due to the lack of dopamine, the levodopa dosage was increased to 60 mg/day. The prolactin level returned to normal (9.15 ng/mL), but the abnormal movements persisted. EEG was conducted and during EEG recordings, he experienced habitual seizures in the form of an abrupt loss of awareness and trembling in both hands. Unlike previous tests that had showed normalcy, ictal EEG showed prolonged bursts of 3 to 4 Hz generalized spike and wave discharges (GSW) (Fig. 1). Brain MRI showed diffuse signal increase of deep white matter on T2-weighted and fluid-attenuated inversion recovery images (Fig. 2). Introduction of antiepileptic drugs; valproate 1,000 mg/day, levetiracetam 1,000 mg/day, zonisamide 200 mg/day markedly reduced abnormal movements without adverse events.
This male patient with DHPR deficiency, presented epileptic seizures at age 3 months. By EEG evaluation of ictal epoch, we could document that his epileptic seizures were idiopathic generalized epilepsy, especially juvenile absence epilepsy, since he experienced limb impaired awareness, myoclonic jerks related to EEG GSW discharges.
We also demonstrated that antiepileptic drugs such as valproate, levetiracetam and zonisamide should be introduced to suppress his seizures; conversely, a dosage increase of neurotransmitter supplementation was not effective.
The present case, wherein the treatment including neurotransmitter supplementation was started when the patient was age seven, resulted in severe epileptic seizures. In patients with drug-resistant epilepsy, or in infants with new-onset epilepsy in whom the initial testing did not reveal an underlying etiology, a full metabolic workup including hyperphenylalaninemia, is needed.
Conversely, if the abnormal movements persist in the DHPR deficiency, it is necessary to assess if another condition such as epilepsy co-exists. A previous paper described that generalized seizures have been reported in 25% of hyperphenylalaninemia [3], and the association between hyperphenylalaninemia and seizures is not well known [4]. Patients with normal EEG in infancy may have abnormal EEG findings when retested later, even if they maintain their treatment [5].
Serial evaluation of EEG, brain MRI is essential in DHPR deficiency patients with unusual movements even though those movements are undefined and the previous test was normal. Early detection of epilepsy can change the patientŌĆÖs prognosis by reducing the frequency of seizures and prevent developmental delay.