Febrile Infection-Related Epilepsy Syndrome: Refractory Status Epilepticus and Management Strategies
Article information

Abstract
Febrile infection-related epilepsy syndrome (FIRES) is a rare, catastrophic epileptic syndrome that strikes previously healthy children. Its pathogenesis is unknown, it has few treatments, and it is typically refractory. In FIRES, refractory status epilepticus or a cluster of seizures starts a few days after the onset of an acute febrile illness, and it may continue as drug-resistant epilepsy, with neuropsychological impairments occurring without latency. Clinical knowledge and guidelines on FIRES are limited because it is sporadic and extremely rare. To date, the absence of specific biomarkers poses a significant diagnostic challenge; nevertheless, early diagnosis is very important for optimal management. Despite treatment with multiple immunotherapies and anti-seizure medications, the majority of patients with FIRES are left with significant cognitive disabilities and refractory epilepsy. This review aims to highlight the most recent insights into the clinical features, terminology, epidemiology, pathogenesis, diagnostic challenges, and therapeutic options associated with FIRES.
Introduction
The acronym febrile infection-related epilepsy syndrome (FIRES) was first used by van Baalen et al. [1] in 2010, although the same clinical entity has been given different names by other authors [2]. If infectious encephalitis has been excluded, FIRES should be suspected whenever a previously healthy child presents with the explosive onset of severe status epilepticus (SE) refractory to even anesthetics, shortly after a brief, often convalescing, febrile illness [1]. The term refers to a rare disastrous epileptic encephalopathy with a yet undefined etiology, characterized by acute manifestation of recurrent seizures or refractory SE preceding febrile illness, but without evidence of infectious encephalitis.
In general, the etiology of FIRES is unclear, and alternative hypotheses of epileptogenesis exist. Despite the absence of biomarkers, early diagnosis is very important for optimal management as diagnostic and therapeutic delays contribute to poor outcomes following SE. In up to 60% of cases of de novo refractory SE, diagnostic work-up fails to identify the underlying etiology, which is the typical and problematic clinical situation associated with FIRES [3]. Because FIRES is extremely rare, this sudden and severe epileptic encephalopathy is challenging to clinicians. This clinical review focuses on correctly recognizing and managing FIRES.
Terminology
A number of names and acronyms have been proposed to describe the syndromes here referred to as FIRES, including new-onset refractory status epilepticus (NORSE), acute encephalitis with refractory repetitive partial seizures (AERRPS), and devastating epileptic encephalopathy in school-aged children (DESC) [4]. Although described in adults, the clinical manifestations, course, and outcomes of NORSE are very similar to FIRES. Initially, NORSE was considered to be specific to the adult population while FIRES was specific of the pediatric population; however, current scientific evidence does not support differentiating the two syndromes [5]. The recently proposed consensus definition identifies FIRES as a subcategory of NORSE [5]. The consensus is that NORSE describes a clinical presentation, but is not a specific diagnosis, in a patient without active epilepsy or other existing relevant neurological disorder who experiences a new-onset of refractory SE in the absence of a clear acute or active structural, metabolic, or toxic cause. FIRES, as a subtype of NORSE requires a prior febrile infection, with fever starting between 24 hours and two weeks prior to the onset of refractory SE. Fever may or may not be present at the onset of SE, and patients may be of any age. This definition excludes prolonged febrile seizures, which usually occur in children who have had a new-onset of fever <24 hours prior to the onset of seizures, or whose fever is recognized only after onset of seizures [6].
Epidemiology
Before the term FIRES was accepted, cases of encephalitis-related refractory SE caused by an unknown or assumed immune etiology were described using many different terms; therefore, it is difficult to extract epidemiological data from available literature. Based on a prospective hospital-based surveillance program conducted in Germany, van Baalen et al. [1] estimated the annual incidence and prevalence of FIRES among children and adolescents in Germany to be, 1:1,000,000 and 1:100,000, respectively. Although FIRES also occurs in adults, affected patients are mainly children between 5 and 13 years of age, with cases peaking during school age (between 4 and 9 years) and exhibiting male preponderance [4]. The outcomes associated with FIRES are poor, with a mortality rate of up to 30%, refractory epilepsy following the acute phase, and cognitive delay in 66% to 100% of the survivors [4]. Survivors with previously normal cognition typically develop learning disabilities, and only a minority survive the episode without any neurologic sequelae [4].
Etiology and pathophysiology
The pathogenesis of FIRES is still a matter of debate, with many diverse pathogenic cascades and mechanisms hypothesized. Metabolic, microbiologic, genetic, and immune analyses have not provided a clear mechanism. Mutations in genes associated with fever-sensitive epilepsy, such as sodium voltage-gated channel alpha subunit 1 (SCN1A), DNA polymerase subunit gamma (POLG1), and protocadherin-19 (PCDH19), or in genes associated with infection-triggered encephalopathy and SE are typically absent [5]. Several authors explored the possibility of FIRES being a form of severe infectious encephalitis [4]. However, no published data from brain biopsies of FIRES patients have produced typical findings of encephalitis [1]. It is possible that FIRES is caused by an inflammatory or autoimmune mechanism.
While analysis of cerebrospinal fluid (CSF) from FIRES patients often shows mild pleocytosis, evidence for an infection or a specific autoimmune condition is lacking. Nevertheless, high levels of proinflammatory cytokines/chemokines have been observed in the serum and CSF of patients with FIRES, indicating a role for the immune system in pathogenesis [7]. Sakuma et al. [8] reported a comprehensive study of the inflammatory mediators in children with AERRPS, and observed a detectable upregulation in proinflammatory cytokines/chemokines in the CSF. Changes in cytokines/chemokines levels were more prominent in the CSF than in the serum, suggesting that the inflammation primarily occurs in the central nervous system (CNS). Inflammation seems to contribute to the FIRES cascade through a generalized and ongoing lowering of the seizure threshold [9]. The slow, progressive nature of such a process may explain the delay in FIRES onset following a febrile illness. Progressive accumulation of inflammatory cytokines could then give way to a vicious cycle of aberrant hyperexcitability and/or a structural epileptogenic remodeling of brain networks [10]. Additionally, intrinsic factors, such as a genetic predisposition, may be responsible for the lack of an efficient resolution of the epileptogenic process [11].
The biphasic clinical course associated with FIRES is suggestive of an autoimmune mechanism triggered by an infection. In a few cases, CSF specimens from affected patients show lymphocytic pleocytosis and oligoclonal bands. Patient improvement with immune therapy supports the role of the immune system in the pathophysiology of FIRES [12]. However, the search for specific autoantibodies has so far yielded unconvincing results [2,4].
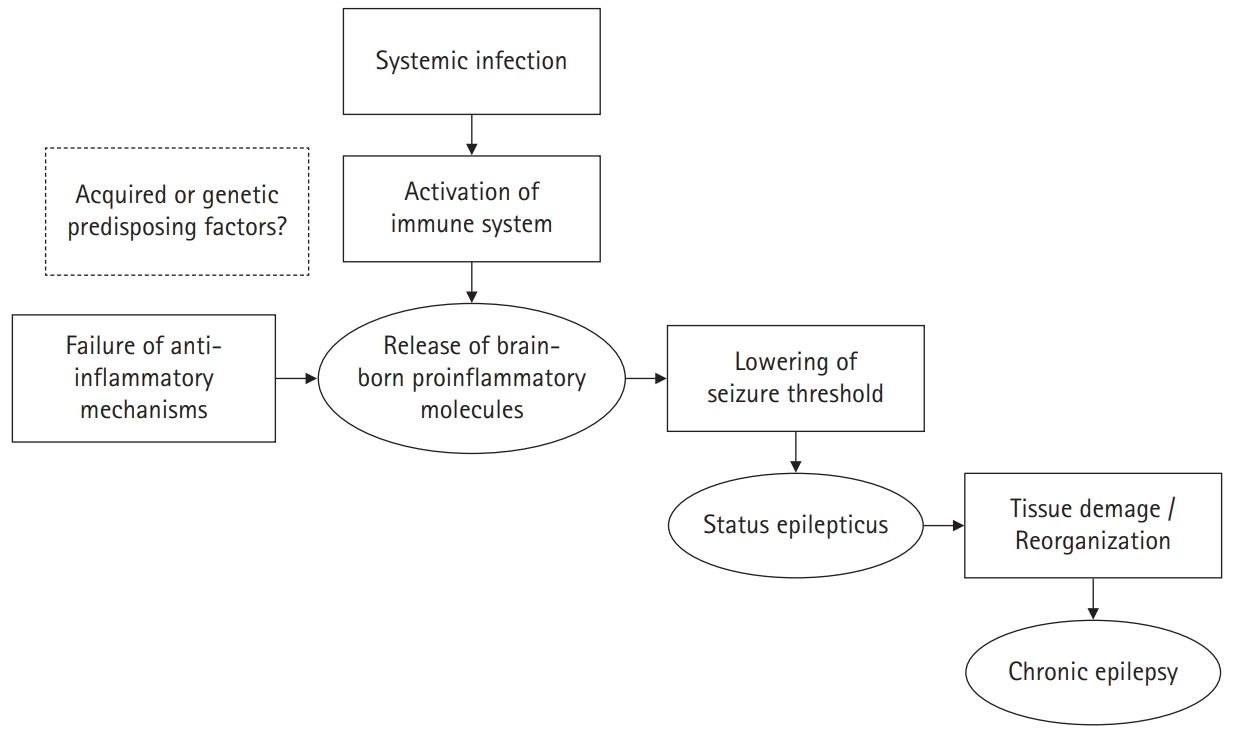
In conclusion, the underlying pathogenic mechanism of FIRES is likely to be a two-hit process, involving the synergistic effect between an immune response to a febrile illness or infection affecting the brain and an intrinsic predisposition toward an auto-sustaining epileptogenic process. The illustration for possible mechanisms of epileptogenesis in FIRES is shown in Fig. 1 [13].
Clinical manifestations
FIRES in previously healthy children generally has three phases. The first phase is the unremarkable febrile illness. The second phase follows between 24 hours and 2 weeks later, with acute, highly recurrent seizures, rapidly evolving into refractory SE. There may or may not be fever at the time of seizure. In approximately 50% of cases, no fever is observed at the time the seizure occurs [14]. Seizures may be focal or multifocal at onset, ranging from dozens to hundreds per day [4,15]. Seizure onset typically presents as a focal seizure, focal seizures with secondary generalization, or secondarily generalized seizures only [4]. Consciousness is impaired during the seizures. Autonomic symptoms, such as pallor, apnea, and cyanosis, have been noted in some patients [15]. SE typically lasts for 1 to 12 weeks, with an average of 3 weeks in duration [16]. The third and chronic phase of FIRES is a drug-resistant epilepsy with neuropsychological and cognitive impairment, occurring without latency as SE decreases [1]. Cluster seizures may occur every 2 to 4 weeks.
Diagnosis
As there is currently no known cause of FIRES, no specific test is available to confirm the diagnosis. Diagnosis depends on clinical assessment ruling out alternative infectious, alternative toxic, metabolic, and genetic etiologies in children with refractory SE in temporal proximity to a febrile illness. By definition, FIRES is a fever-triggered NORSE in patients free from preceding epilepsy or other neurological disorders and without clear or active causes [3]. Therefore, the initial clinical evaluation is crucial and an extensive work-up is necessary to exclude similar conditions associated with acute-onset of SE (Table 1). Careful history-taking, neurological examination, and routine laboratory testing may alternatively identify the most common etiologies of SE, such as discontinuation or changes in antiepileptic drugs (AEDs), intoxication, CNS infections, recent traumatic brain injury, or acute metabolic imbalances. CSF testing in patients with FIRES is usually negative, with mild or no pleocytosis, an absence of markers for viral infections or autoimmunity (oligoclonal band), and an absence of antineural antibodies (anti-voltage-gated potassium channel [VGKC] complex, anti-N‐methyl‐D‐aspartate [NMDA] receptor, and anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] receptor) [17,18].

During the acute phase, standard electroencephalography (EEG) and continuous EEG monitoring are of supreme importance in guiding precise therapeutic interventions, assessing sedation levels, and recognizing nonconvulsive SE [19]. Clinically seizure semiology may indicate focal onset, and EEG ictal recordings can confirm that discharges are often temporal or perisylvian with opercular extension. Interestingly, in a series of published studies, ictal and interictal EEG recordings showed a single epileptic focus in less than 10% of patients [18]. The frequent presence of a shifting focal ictal pattern indicates that both hemispheres are affected while interictal EEG findings show generalized slow waves [17]. Farias-Moeller et al. [20] described the three common features of acute EEG studies in seven patients with FIRES: gradual increase in seizure burden with evolution to SE, recurrent beta-delta complexes resembling extreme delta brush, and a characteristic electrographic seizure pattern, typically beginning with prolonged focal fast activity, followed by well-formed rhythmic spike-and-wave complexes. Unfortunately, EEG has limited value in differentiating FIRES from epilepsy or encephalopathy of autoimmune etiology.
Brain magnetic resonance imaging (MRI) is required to exclude structural abnormalities associated with refractory SE. Initial MRI scans may be negative, or may show abnormalities, predominantly in the temporal regions, insula, or basal ganglia, that may be either transient or irreversible [1,21]. Early MRI scans (within the first weeks) may show swelling in the mesial temporal structures, with increased signal on T2-weighted scans, as observed in autoimmune or limbic encephalitis [1,17]. Follow-up MRI scans (after 6 months) may show bilateral mesial temporal atrophy and increased T2-weighted signal in approximately 50% of cases [17]. Global changes may also occur. Focal abnormal signal changes over the periventricular white matter suggest that more extensive lesions are associated with a poorer clinical outcome [22].
It is important to consider that during childhood, many other conditions can result in prolonged SE, including febrile seizures and febrile SE. Genetic disorders such as Dravet syndrome and Alpers syndrome usually involve fever-associated SE. Therefore, a differential diagnosis should include genetic conditions associated with fever-induced epilepsy, such as in SCN1A-, PCDH19-, or POLG1-related epilepsies (Table 2) [2].

Management
1. Acute phase
The treatment of FIRES represents a significant challenge for clinicians, and is associated with low success rates, especially during the acute phase. There is no evidence of efficacy for any treatment modality except for a ketogenic diet (KD), intravenous immunoglobulin (IVIG), and burst-suppression coma with barbiturates [23]. Multiple different therapeutic options have been reported in small case series demonstrating that no one treatment is superior (Table 3). First-line and second-line immunotherapies, such as high-dose intravenous steroids, IVIGs, and plasmapheresis, may be useful, although efficacy is still debated. The efficacy of tacrolimus, rituximab and/or cyclophosphamide is unclear, although there are some reports of improvement in patients who failed other immunotherapies [18,19,24].

Pediatric medications for febrile infection-related epilepsy syndrome
In most cases, first-line treatment of SE consists of benzodiazepines (lorazepam, diazepam, midazolam, clonazepam), followed by standard anticonvulsant drugs [4]. These drugs are available in intravenous forms, including phenytoin, phenobarbital, levetiracetam, valproic acid, and lacosamide (for focal SE) [25]. Second-line treatments usually require pharmacological coma with an anesthetic infusion of midazolam and barbiturates (pentobarbital in United States and thiopental in Europe) [26]. Midazolam appears as effective as thiopental but is associated with fewer adverse events and better long-term neurological outcomes [27]. High doses of phenobarbital have also been proven effective and elicit fewer side effects than anesthetics [2]. Other anesthetic agents, such as propofol, are used in the management of refractory SE, though its prolonged use in children is discouraged due to the risk of propofol infusion syndrome, a fatal complication occurring when doses of propofol exceed 4 mg/kg/hr for more than 48 hours. Propofol infusion syndrome’s typical features (hypertriglyceridemia, fever, hepatomegaly, or heart failure) are often missing, and other features (arrhythmia, electrocardiographic changes) occur late [28]. Tapering from anesthetics is very difficult and seizure recurrence is common. Moreover, there are concerns regarding anesthetics’ use in FIRES, since prolonged burst-suppression coma has a significant association with a worse cognitive outcome and with a more severe course of disease [2]. Medications with an intravenous (IV) formation are favored, but topiramate, pregabalin, and/or clobazam are sometimes used later as add-on therapy.
The KD has been used successfully in patients with refractory seizures and, according to some reports, has shown some efficacy in FIRES (Table 4) [4,29,30]. It has been hypothesized that the KD may not only have an anticonvulsant effect (e.g., through the production of decanoic acid, which induces a direct inhibition of the post-synaptic excitatory AMPA) but also an anti-inflammatory effect. Early introduction of a KD in children with FIRES could be effective not only during the acute phase but also in long-term epilepsy management and cognitive outcomes [2]. It is hoped that future prospective controlled studies can confirm the efficacy of KD in the treatment of FIRES.

Previous literature on the therapeutic effects of KD in patients with FIRES
Because inflammation is presumed to play a causal role in FIRES, approaches that modulate the immune system have been employed. However, these treatments have not been systematically studied. High-dose intravenous steroids are often used [31]; however, this treatment is associated with significant adverse effects. Treatment with IVIG is being explored as an option, but its efficacy has not been demonstrated [32]. The efficacy of plasmapheresis has also not been demonstrated [32]. Other agents, such as anakinra, a recombinant, modified version of the human interleukin-1 receptor antagonist protein, have been effective in some patients, both in the acute phase and in the prevention of adverse outcomes [33]. Tocilizumab is a humanized monoclonal antibody against the interleukin-6 receptor, and currently used for various autoimmune diseases, including rheumatoid arthritis, systemic juvenile idiopathic arthritis, and antibody-mediated encephalitis. Recently, Jun et al. [34] reported the therapeutic effectiveness of tocilizumab in the patients with NORSE who remained refractory to conventional immunotherapy [35].
Cannabidiol (CBD) works by decreasing glutamate and gamma-aminobutyric acid synaptic transmission in the brain. The resultant decrease in excitatory neurotransmitter release may increase the seizure threshold [36]. In a recently published case series, CBD appeared effective in reducing the frequency and duration of seizures in patients with FIRES who had not responded to standard AEDs or other therapies [37]. Magnesium infusions are generally not efficacious, but there are isolated reports of good seizure control in children with FIRES after continuous intravenous infusion of magnesium sulfate [38]. In these children, serum magnesium concentrations ranging from 2.1 to 5 mmol/L were achieved. No significant adverse effects were observed.
Therapeutic hypothermia may also be effective in the control of SE by reducing proinflammatory cytokine levels and protecting the integrity of the blood-brain barrier [5]. Preliminary data showed that therapeutic hypothermia at 33°C may be effective in the control of SE [5]. Vagus nerve stimulation (VNS) is typically introduced as long-term treatment for seizure control after recovery from SE. A recent retrospective review reported the effect of VNS in controlling SE in children, and found that VNS appeared to have a favorable effect on SE, NORSE and generalized convulsive seizures in children with post-FIRES medically intractable epilepsy [39].
2. Chronic phase
No systematic study exists for treatment of FIRES in the chronic phase. Despite polytherapy, the severity and frequency of seizures increase intermittently and during infections, again resulting in SE. If FIRES is inflammation-mediated, the anti-inflammatory effects of CBD or anakinra may have anti-seizure effects not only in the acute but also in the chronic phase of illness [37]. Additionally, we found that KD, clobazam, and phenobarbital are often employed as therapeutic options during the chronic phase [40].
Prognosis
The prognosis following FIRES is poor. The seizures are recalcitrant, prolonged, and difficult to control [41]. No therapeutic modality is efficacious in shortening the acute phase, with the exception of KD. Outcomes appear to be better when the KD is used early and if KD is effective in producing better outcomes. Therefore, the prognosis may improve due to both earlier diagnosis and earlier KD [42]. Mortality associated with FIRES is 10% during the acute phase and 13% during the chronic phase [7]. Only one-third of the surviving patients had normal or borderline cognitive level; one-third had mild-to-moderate intellectual disability, and one-third had severe mental retardation or were in a vegetative state. Nearly all patients had refractory epilepsy at follow-up. Cognitive levels at follow-up were significantly related to the duration of burst-suppression coma and younger age at onset of FIRES [43]. The presence of higher signal periventricular white matter lesions may be correlated with poorer neurological outcomes [7]. However, very few long-term observations exist to date.
Conclusions
FIRES is a rare epilepsy syndrome of unclear etiology in which children, usually of school age, suddenly develop very frequent seizures after a mild febrile illness. Seizures in FIRES are notoriously difficult to treat. The majority of children with FIRES are left with significant cognitive disability and refractory epilepsy. Treatments have poor efficacy, and therefore new insights concerning FIRES pathogenesis are still desperately needed in order to develop more targeted therapies.
Notes
No potential conflict of interest relevant to this article was reported.
Authors’ contributions
Conceptualization: YJL. Data curation: YJL. Formal analysis: YJL. Project administration: YJL. Visualization: YJL. Writing-original draft: YJL. Writing-review & editing: YJL.