Introduction
With the advent of next-generation sequencing, there have been significant advances in the genetics of epilepsy within the last decade [1]. In developmental and epileptic encephalopathy patients, targeted gene panels and/or whole exome sequencing have now become part of the routine diagnostic workup, which provide a genetic diagnosis in about 30% of patients.
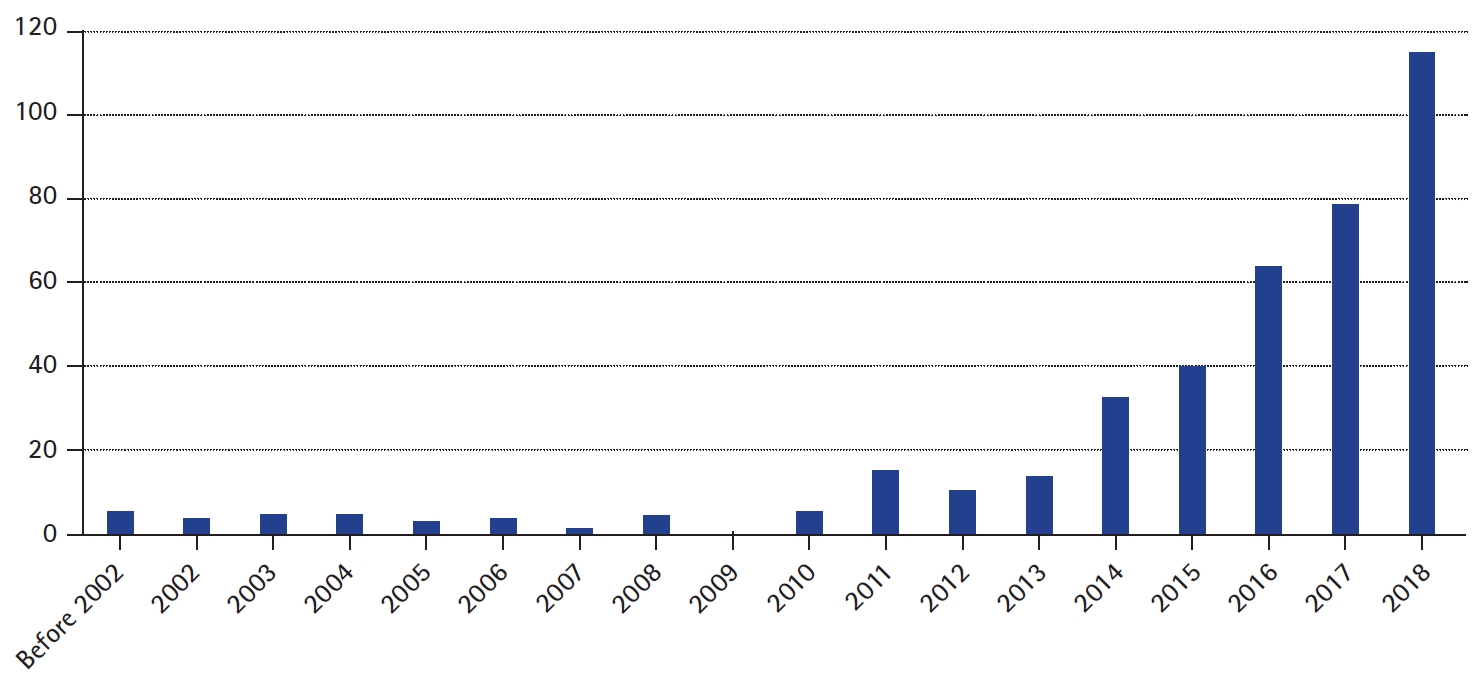
As the number of genetically diagnosed patients has increased, so has the demand for more precise treatment based on pathogenic genetic mutations. The demand has resulted in active research and a rapidly increasing number of publications focused on precision medicine in epilepsy (Fig. 1) [2]. Based on current knowledge, approximately 25% of genetically diagnosed epilepsy patients with de novo monogenic mutations carry potential targets for precision medicine approaches [3].
In this article, we review current knowledge regarding gene-specific therapies for some forms of developmental and epileptic encephalopathy caused by genes with high diagnostic yields, which are therefore, more frequently encountered by treating physicians.
ALDH7A1
The aldehyde dehydrogenase 7 family, member A1 (ALDH7A1) gene encodes the enzyme ╬▒-aminoadipic semialdehyde (╬▒-AASA) dehydrogenase, also known as antiquitin, a key enzyme in lysine metabolism [4]. Biallelic mutations of ALDH7A1 result in deficiency of ╬▒-AASA dehydrogenase, which then results in the accumulation of ╬▒-AASA and the cyclic equivalent ╬ö-1-piperideine-6 carboxylate (╬ö1-P6C) [4]. The accumulated ╬ö1-P6C sequesters the active form of vitamin B6 (pyridoxal 5ŌĆ▓-phosphate [PLP]), and causes pyridoxine-dependent epilepsy [4].
The classic presentation of pyridoxine-dependent epilepsy is the neonatal onset of treatment-resistant seizures that respond dramatically, both clinically and electroencephalographically, to pyridoxine supplementation [5]. Despite adequate seizure control achieved with pyridoxine alone, 75% of patients with pyridoxine-dependent epilepsy have significant intellectual disability and/or developmental delay [6]. The degree of intellectual disability or developmental delay does not correlate with seizure control or the age at which pyridoxine treatment is initiated [6].
Treatment with pyridoxine supplementation in pyridoxine-dependent epilepsy patients is therapeutic because it overcomes the secondary depletion of pyridoxal phosphate (PLP) [5]. Pyridoxine supplementation alone does not treat the underlying defect of lysine oxidation, but treatment with a lysine-restricted diet and/or with a lysine transport inhibitor (arginine) has been shown to improve cognitive function at some level [7-9]. Triple therapy with pyridoxine, arginine supplementation, and dietary lysine restriction has been recommended to ameliorate the cognitive impairment seen in pyridoxine-dependent epilepsy [9].
CDKL5
Cyclin-dependent kinase-like 5 (CDKL5) encephalopathy is a severe X-linked neurodevelopmental disease caused by mutations in the CDKL5 gene, which lead to deficiency in CDKL5 protein expression or function [10]. CDKL5 is highly expressed in the brain, mainly in neurons, with both nuclear and dendritic localization, and its expression peaks during the postnatal period [11,12]. In mouse models, Cdlk5 knockout animals exhibited hippocampus-dependent learning and memory impairment, visual and respiratory deficits, and motor stereotypes, which were associated with neuroanatomical alterations such as reduced dendritic branching of hippocampal and cortical neurons, reduced dendritic spine density, and altered connectivity [13-19].
Epilepsy in CDKL5 encephalopathy patients often starts before 2 months of age, with frequent generalized tonic seizures that typically last for less than 1 minute [20]. Seizures at this time are well-controlled with antiepileptic drugs (AEDs), and electroencephalograms (EEGs) are usually normal [20]. Epilepsy relapses in late infancy at a median age of 11 months, when infantile spasms with hypsarrhythmia, sometimes combined with brief tonic medication-resistant seizures, emerge [20]. During the year after onset of infantile spasms, epilepsy evolves into late multifocal and myoclonic epilepsy, showing frequent pharmacoresistant seizures of multiple types such as myoclonic seizures, and EEGs showing high-amplitude delta waves with pseudo-periodic bursts of high-amplitude spikes [20]. Severe neurodevelopmental delay is present from the beginning, but there is usually no period of regression [21].
Seizures in CDKL5 encephalopathy, especially seizures during the ŌĆ£late epilepsyŌĆØ stage are highly refractory, and there are no available AEDs that are effective in controlling these seizures. A promising study regarding cannabidiol came out in 2018, which analyzed patients with treatment-resistant, childhood-onset epilepsies including CDKL5 encephalopathy [22]. In CDKL5 encephalopathy patients (n=17), 50% responder rate was 41% at week 12, and 53% by week 48 [22]. However, cautious interpretation is needed due to the small number of subjects and due to exclusion of withdrawn patients from analysis. Therefore, additional placebo-controlled randomized trials with larger sample sizes are needed.
Several labs are assessing the use of drugs such as tianeptine and tideglusib in CDKL5 encephalopathy. Tianeptine is an antidepressant that has been used for more than 30 years. Studies utilizing neurons from Cdkl5 knockout mouse models have proposed that CDKL5 deficiency in primary hippocampal neurons negatively affects the expression of the alpha-amino-3-hydroxy-5-methyl-4-iso-xazole propionic acid (AMPA) receptors, and such an effect is likely to contribute, at least in part, to the altered synaptic functions and cognitive impairment linked to loss of CDKL5 [23]. Therefore, tianeptine, which is known to recruit and stabilize AMPA receptors at the synaptic sites, has been studied in vitro and results revealed that tianeptine normalized the expression of membrane-inserted AMPA receptors [23].
Tideglusib is a glycogen synthase kinase 3 beta (GSK3╬▓) inhibitor, and is currently undergoing clinical trials for use against diseases such as AlzheimerŌĆÖs, progressive supranuclear palsy, and myotonic dystrophy. In mouse, deficiency of Cdkl5 caused defects in postnatal hippocampal development and hippocampus-dependent learning and memory. These defects were accompanied by increased activity of GSK3╬▓, an important inhibitory regulator of many neuronal functions [17]. Therefore, tideglusib was tested on Cdkl5 knockout mice and results showed that tideglusib improved hippocampal development, hippocampus-dependent behaviors, and memory performance in juvenile Cdkl5 knockout mice [24].
Finally, CDKL5 protein substitution therapy was successfully conducted in a mouse model using a protein transduction domain (TAT) that was able to deliver macromolecules into cells, and even into the brain when fused to a specific protein [25]. Intracerebroventricular infusion of TAT-CDKL5 protein was efficiently internalized by target cells, which resulted in the retention of CDKL5 activity and the restoration of hippocampal development, hippocampus-dependent memory, and breathing pattern in Cdkl5-null mice [25]. Systemically administered TAT-CDKL5 protein also crossed the blood-brain barrier, reached the central nervous system, and improved behavioral defects in mice [25].
KCNQ2
Mutations in the potassium channel, voltage-gated, KQT-like subfamily member 2 (KCNQ2) gene, encoding voltage-gated K+ channel subunits underlying the neuronal M-current, usually lead to loss-of-function of KV7.2 and cause a decrease in neuronal M-current conductance, thereby increasing neuronal excitability [26]. Functional studies have revealed that a 25% reduction in KV7.2 current is sufficient to increase neuronal excitability to epileptogenic levels in early infancy [27]. However in rare cases, variants of KCNQ2 such as R144Q, R198Q, R201C, and R201H, produce gain-of-function effects and are reported to be associated with early-onset epileptic encephalopathy, infantile spasms, or neonatal nonepileptic myoclonus [28-30].
Benign familial seizures of early infancy and early infantile epileptic encephalopathy (EIEE) are two main types of epilepsy associated with KCNQ2 mutations. Seizure onset in both syndromes occurs as early as in the first days of life [31]. Patients with benign seizures in early infancy have an excellent prognosis regarding both seizure remission and neurodevelopment. In contrast, patients with EIEE suffer from a severe phenotype comprising drug-resistant seizures, intellectual disability, and encephalopathic EEG [31].
A systematic review conducted in 2019 analyzed the medications used in EIEE, and revealed that sodium channel blockers were the most frequently used monotherapy agent [31]. The distribution of AEDs used by patients achieving control of their seizures was 37.8% with sodium channel blockers, 26.3% with valproic acid, and 33.3% with levetiracetam [31]. Treatment trials with phenobarbital were largely unsuccessful: 8.6% of the trials led to seizure freedom in patients while 74.3% of the trials showed no effect [31]. Therapeutic response to sodium-channel blockers in patients with KCNQ2 mutations could be explained by the fact that voltage-gated sodium channels and KCNQ potassium channels co-localize at critical locations on the neuronal membrane and therefore, modulation of one channel may significantly affect the function of the channel complex [32,33]. Because of this, sodium channel blockers such as phenytoin, carbamazepine, and oxcarbazepine are recommended as a first-line treatment [31,34]. However, sodium channel blockers were not clearly superior to levetiracetam or valproate, indicating that the latter AEDs might be considered in patients who failed to respond to sodium channel blockers [31].
Retigabine (also known as ezogabine) is a Kv7.2/Kv7.3 opener that leads to neuronal hyperpolarization of the membrane potential and was first introduced as an adjunctive therapy in adults with focal seizures [35]. Despite its effectiveness, retigabine was withdrawn from the market due to lack of demand caused mainly by side effects such as blue discoloration of skin and retina [36]. However, the fact that retigabine opens the Kv7 potassium channel drew interest in its use for patients with KCNQ2 mutations, and the results were favorable in animal models [37]. Of all mono-therapeutic treatment trials with retigabine, only 14.3% led to seizure freedom while 71.4% showed no effect and current findings do not support use of retigabine compared to other drugs such as sodium channel blockers [31].
KCNT1
The potassium channel subfamily T member 1 (KCNT1) gene encodes the sodium-activated potassium channel KCa4.1. KCNT1 is widely expressed throughout the brain, as well as in the dorsal root ganglia, kidney, and heart, and is responsible for slow hyperpolarization after bursts of action potentials [38,39]. The pathogenic KCNT1 mutations described to date are all missense variants; no nonsense or other truncating mutations have been identified yet [40]. This suggests that perturbation of normal KCNT1 protein function, rather than the loss of its function, is the underlying pathomechanism [40]. This is consistent with other reports, including animal models demonstrating that KCNT1 pathogenic variants manifest a gain-of-function effect with increased current amplitude [39,41,42].
Mutations in KCNT1 cause a wide spectrum of epileptic disorders with variable ages at onset and cognitive outcomes including Ohtahara syndrome, West syndrome, and severe nocturnal frontal lobe epilepsy (NFLE). However, epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) are the two most characteristic epilepsies associated with KCNT1 [39,41,43,44]. KCNT1 is the most frequently identified genetic cause of EIMFS, and 39% (28 out of 71) of individuals from EIMFS patient cohorts that have been analyzed for KCNT1 mutations are mutation positive [40]. Patients with ADNFLE and NFLE with KCNT1 mutations have a more severe phenotype than ADNFLE patients with mutations in nicotinic acetylcholine receptor subunit genes in terms of earlier age of seizure onset, marked increase in frequency of refractory seizures, and high frequency of comorbid intellectual disability and psychiatric features [44].
Quinidine is an inhibitor of several types of potassium channels, including KCNT1, and is currently used as an antiarrhythmic agent and antimalarial drug [40,45,46]. Given that functional studies have shown mutations in KCNT1 cause an increase in channel current, quinidine gathered interest as a potential inhibitor that could reverse this abnormal channel function and therefore, treat seizures in patients with mutations in KCNT1 [41,42,47,48]. Indeed, the effectiveness of quinidine in reversing the increased current of mutant KCNT1 channels has been demonstrated in vitro [42,48]. However, in an order-randomized, blinded, placebo-controlled, crossover trial of quinidine in six ADNFLE patients, 33.3% (n=2) of patients discontinued therapy due to prolonged QT interval occurring at serum quinidine levels below the therapeutic range, and none of four patients who completed treatment trial showed 50% seizure reduction [49]. Other reports that included 25 EIMFS, two focal epilepsy, one West syndrome, and one NFLE patients, demonstrated that 31.0% (n=9, including eight EIMFS) of patients experienced improvements in seizure activity due to quinidine administration while the others did not show any improvements [39,48,50-60]. Lack of therapeutic response observed in the majority of patients is likely because exposure levels in the brain are too low to cause significant in vivo channel blockade [52].
SCN2A
The sodium channel, voltage-gated, type II, alpha subunit (SCN2A) gene encodes the neuronal sodium channel NaV1.2, one of four sodium channel paralogs expressed throughout the central nervous system, along with NaV1.1 (SCN1A), NaV1.3 (SCN3A), and NaV1.6 (sodium channel, voltage-gated, type VIII alpha subunit [SCN8A]) [61]. Mutations in SCN2A are primarily associated with three distinct disorders: (1) infantile epileptic encephalopathy characterized by drug-resistant seizures with age of onset less than 12 months, followed by neurodevelopmental delay; (2) benign (familial) infantile seizures characterized by seizure onset at less than 12 months of age, which resolve by 2 years of age without overt long-term neuropsychiatric consequences; and (3) autistic spectrum disorder/intellectual disability (ASD/ID) characterized by global developmental delay, particularly of social and language milestones, with up to one-third of children developing childhood-onset seizures after 12 months of age [61-63].
An integrated analysis of genetic and electrophysiological data suggested a model explaining the three disorders associated with SCN2A (Fig. 2) [61]. Variants associated with gain-of-function of NaV1.2 channel activity led to infantile epileptic encephalopathy and benign infantile seizures, while those associated with loss-of-function led to ASD/ID [62,63]. Further, the degree to which the gain-of-function variants potentiate NaV1.2 activity distinguishes infantile epileptic encephalopathy from benign infantile seizures, with severe variants leading to infantile epileptic encephalopathy [62,63]. This model is supported by several other findings including observations using in vitro electrophysiology, the restriction of protein truncating variants resulting in loss of function in ASD/ID cases, shared symptoms between individuals with recurrent missense variants, and the clustering of infantile epileptic encephalopathy/benign infantile seizure variants around the voltage sensor domain of the channel while the ASD/ID missense variants cluster near the pore loop regions [62,63].
Seizures in SCN2A encephalopathy are often resistant to AEDs. However, for infants less than 4 months of age, sodium channel blockers such as phenytoin and carbamazepine are more effective, which is expected considering the gain-of-function nature of causative mutations [62,63]. For children more than 12 months of age with ASD/ID and childhood-onset seizures, the opposite action should be taken, and drugs other than sodium channel blockers, such as levetiracetam, benzodiazepines, and valproate, are therapeutic options. New emerging therapies such as bromides, fenfluramine, and cannabidiol are being studied and have shown efficacy in treating Dravet syndrome [64]. Whether such medications would provide benefit with SCN2A associated late-onset seizures remains unknown. However, when considering such treatments, it is important to remember that the NaV1.1 channels affected in Dravet syndrome are more commonly expressed in inhibitory neurons, and that loss-of-function of NaV1.1 may have opposing effects in brain networks compared with NaV1.2 loss-of-function in excitatory neurons [65].
SCN8A
The SCN8A gene encodes the pore-forming voltage-gated sodium channel subunit Nav1.6, which is widely expressed in the brain and is responsible for the initiation and propagation of neuronal action potentials contributing to regulation of neuronal excitability [66-68]. Most mutations identified in SCN8A result in gain-of-function changes in biophysical properties leading to elevated channel activity, either due to premature channel opening or impaired channel inactivation [69-73]. However, a small subset of mutations cause loss of function of SCN8A, resulting in isolated intellectual disability, myoclonus, and movement disorders [74,75].
SCN8A encephalopathy patients usually show severe developmental delay and intellectual disability, pharmacoresistant epilepsy with seizures starting before 18 months of age, in addition to pyramidal and extrapyramidal signs [76,77]. Epilepsy in SCN8A encephalopathy patients comprises multiple seizure types, including focal, generalized, and epileptic spasms [78]. Sudden unexpected death in epilepsy is reported in approximately 10% of cases [77].
As SCN8A epileptic encephalopathy is mostly due to gain-of-function mutations of SCN8A, the most effective AEDs reported so far are sodium channel blockers including phenytoin, carbamazepine, and oxcarbazepine, usually at supratherapeutic doses [77]. Also, at least one study reported improvements in seizure clusters and non-convulsive status epilepticus when patients were treated with benzodiazepines [77]. Of note, levetiracetam was reported to be ineffective and may even worsen seizure activity [77].
STXBP1
The syntaxin-binding protein 1 (STXBP1) gene encodes the STXBP1 protein, which is an essential protein for presynaptic vesicle docking and fusion through interaction with the soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) complex proteins, and has an essential function in neurotransmitter release [79,80]. STXBP1 encephalopathy is caused by both truncating and missense mutations in STXBP1, and the current leading hypothesis is that STXBP1 loss-of-function is the common pathomechanism, although some in vitro studies have reported possible dominant-negative effects or gain in pathological function [80-83].
STXBP1 encephalopathy patients show developmental delay/intellectual disability, epilepsy, autism spectrum disorders, and involuntary movements such as spasms and jerks [84,85]. Epilepsy in STXBP1 encephalopathy patients is characterized by early onset of tonic spasms, often immediately after birth, and drug-resistant seizures with multifocal epileptic activity on EEG [86].
Some case reports are currently available, and some beneficial AEDs include vigabatrin, valproic acid, and levetiracetam [81,82,87-91]. A beneficial effect of levetiracetam is noteworthy because it has been proposed to modulate synaptic vesicle release via binding with synaptic vesicle glycoprotein 2a [80,92]. However, there is insufficient data for this generalization. There is currently no drug on the market that directly targets STXBP1.
SYNGAP1
The synaptic RAS-GTPase-activating protein 1 (SYNGAP1) gene encodes SYNGAP1 protein, which is an important mediator in the N-methyl-D-aspartate (NMDA) receptor-activated RAS-signaling cascade that regulates postsynaptic density and formation, development, and maturation of dendritic spines [93,94]. In a mouse model, heterozygous mutations in Syngap1 led to protein truncations and abnormal dendritic spines during development [95], decreasing the ability of Syngap1 to bind downstream molecules in the signaling pathway, and thus failing to inhibit Ras activity. Increased Ras activation led to the activation of molecules that regulate the actin cytoskeleton, thus shifting the equilibrium towards the more stable actin form [93,96]. This, combined with an increase in insertion of AMPA receptors into the postsynaptic membrane during development, caused the dendritic spines to mature into mushroom-shaped spines earlier than normal, leading to elevated excitatory synaptic transmission and excitatory/inhibitory imbalance. The occurrence of these events during a critical period of development made the neurons more prone to seizures [93,97].
SYNGAP1 mutations cause developmental epileptic encephalopathy characterized by generalized epilepsy with eyelid myoclonia with absences and myoclonic-atonic seizures that are often pharmacoresistant [98]. Seizures triggered by eating is also a characteristic feature [98]. SYNGAP1 encephalopathy is associated with a spectrum of mild to severe intellectual disability, with a large proportion of patients with severe ID and other comorbidities such as behavioral problems, autism spectrum disorder, hypotonia, eating problems, and sleeping problems [98].
In a retrospective study with 57 patients, valproate (n=45) and lamotrigine (n=22) were the most commonly prescribed AEDs, with long-term treatment with lamotrigine in 77% and valproate in 64% of patients, respectively, suggesting effectiveness [98]. This is a reasonable result considering that generalized seizures occur in SYNGAP1-associated epilepsy patients and because there is currently no targeted therapy specifically for SYNGAP1.
Conclusion
Here, we have reviewed several developmental and epileptic encephalopathies caused by frequently identified genes. Already too well known SCN1A was not addressed. Dietary therapy and modulatory therapy such as vagus nerve stimulation were also not covered in this review. Therapies directly targeting at causative mutations, such as retigabine in KCNQ2 encephalopathy and quinidine in KCNT1 encephalopathy were discussed. However, despite promising results in vitro, the results were disappointing when administered to patients. Considering the pathogenic mechanisms of causative mutations, sodium channel blockers are recommended for patients with KCNQ2 mutations, infantile epileptic encephalopathy patients with SCN2A mutations, and patients with SCN8A mutations. Levetiracetam can be considered in patients with STXBP1 mutations (Table 1).
With a rapidly increasing number of genetically diagnosed patients, development of gene-specific therapies is in great demand. Studies to identify personalized therapeutic strategies based on functional studies of individuals will increasingly be required to meet this demand.