Charcot-Marie-Tooth disease (CMT) is a genetically and phenotypically heterogeneous group of disorders [1]. CMT is divided into types based on clinical characteristics and the associated genes. These types share common clinical features, such as atrophy and weakness predominantly affecting the distal muscles, as well as foot deformities [2]. The two major types, CMT1 and CMT2, can be distinguished by nerve conduction velocity (NCV) results. Genetic testing for CMT became possible after the discovery of the peripheral myelin protein 22 (PMP22) duplication in 1991 [3], which causes the majority of CMT1 cases [4]. PMP22 duplication is examined in the first screening; however, the genetic cause of more than 50% of clinically diagnosed CMT cases remains unknown [5]. Rapid progress in gene discovery has been attributed to the development of multiple-parallel or next-generation sequencing (NGS) technology, which has improved genetic diagnoses. Here, we describe three patients who were diagnosed with CMT using the PMP22 duplication test and/or NGS.
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of Yeungnam University Hospital (YUMC 2019-08-077). All patients or their legal guardians provided their written informed consent for these cases to be reported here.
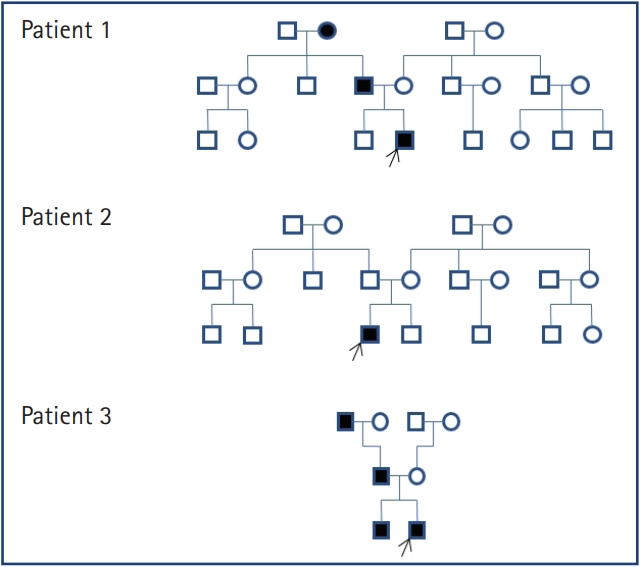
Patient 1: A 16-year-old Korean boy attended our hospital with a gait disturbance and hand tremor. He walked on his toes and had essential tremor of the hand. On physical examination, he had high arches and his ankles pointed down. His father and brother reported experiencing pain when walking; they were suspected to have CMT. The patient underwent nerve conduction studies (NCSs), which revealed decreased conduction velocity (CV) in the lower extremities, a characteristic pattern of demyelinating disease that distinguishes it from axonal disease. Based on his NCS findings and family history of CMT, the patient was screened for PMP22 duplication and tested positive. He was diagnosed with CMT1A inherited in an autosomal dominant fashion. His father was also diagnosed with CMT1A; however, his brother was negative for the mutation. The patient is currently undergoing physical therapy to improve his symptoms.
Patient 2: A 14-year-old boy presented with a gait disturbance. His mother reported that his gait had changed recently; however, the patient was not aware of it himself. There was no family history or symptoms of CMT, but he had several clinical signs of CMT, including ankle drop, pes cavus, and a steppage gait. NCS revealed a marked decrease in CV. Considering his symptoms, CMT was strongly suspected, but the PMP22 test was negative. NGS was performed to determine whether the patient had another type of CMT. This revealed a gap junction beta 1 protein (GJB1) gene mutation (c.109-G>T), which is associated with CMTX1. CMTX1 is an X-linked dominant form that is the second most common form of CMT. Since his diagnosis, this patient has received joint follow-up with orthopedic surgery and rehabilitation medicine.
Patient 3: A 19-year-old Korean man presented with bilateral ankle pain. The pain had become worse over time, and after 1 year the patient experienced pain while standing. He had a high-arched foot deformity, and his grandfather, father, and brother had similar symptoms. Considering the family history, his father requested an examination via NGS. The NGS findings revealed an uncommon type of CMT. This mutation involved the leucine-rich repeat and sterile alpha motif-containing 1 (LRSAM1) gene; the c.2008G>T mutation of the LRSAM1 gene is very rare and may cause CMT2P in an autosomal dominant or recessive genetic pattern.
CMT (also known as hereditary motor and sensory neuropathy) is one of the most common inherited neuromuscular diseases [6]. CMT is associated with length-dependent degeneration of the axon or myelin sheath of the peripheral nerves. It is characterized by progressive motor weakness, sensory abnormalities, and abnormal NCV or amplitude. CMT is classified into three clinical subtypes according to the NCV findings: demyelinating, axonal, and intermediate. It is further divided into several genetic subtypes, including CMT3, CMT4, CMT5, CMT6, and X-linked CMT [7].
An understanding of the genetics of CMT began in 1991 with the discovery of a duplication in the short arm of chromosome 17, which encompasses the PMP22 gene. Duplication of PMP22 is the most common cause of CMT, and this type of CMT is classified as CMT1A. The three most common genetic causes are mutations in GJB1, myelin protein zero (MPZ), and mitofusins 2 (MFN2), which constitute the majority of additional results from current genetic testing [4]. NGS can rapidly decipher vast amounts of genomic information by decomposing one genome into many fragments, reading each fragment simultaneously, and combining the fragments via bioinformatics techniques. The NGS performed at our hospital can test 52 related genes, including PMP22. Because of rapidly evolving NGS technology and decreasing costs, NGS is becoming a practical and affordable technique for testing a large set of genes [5].
A list of patients diagnosed with CMT and their pedigrees are shown in Table 1 and Fig. 1. We performed the PMP22 duplication test in patient 1, who was diagnosed with CMT1A. Patient 2 had a negative PMP22 result; NGS identified a GJB1 mutation, which is associated with CMTX1. The GJB1 mutation causes a malfunction of gap junctions and interferes with communication between Schwann cells, thereby impeding the transmission of nerve impulses. Patient 3 underwent NGS only, and an LRSAM1 mutation related to CMT2P was identified. LRSMA1 is a ubiquitin ligase and its mutation is well expressed in axonal CMT2P. However, it remains unknown how this leads to CMT.
Among individuals with foot deformities, 50% to 80% of cases are caused by CMT, particularly when they are bilateral [8]. Clinicians should therefore consider CMT when treating children with foot deformities. Currently, treatment for CMT is focused on maintaining joint and muscle function [9]. Although there is no cure for CMT, a precise and early diagnosis can facilitate better counseling, and early rehabilitation can improve quality of life [10]. Additional advances in genetic testing are necessary.
In conclusion, although the various types of CMT share common clinical features, CMT is a genetically heterogeneous group of disorders. NGS can detect PMP22 mutations and other genomic mutations, and it is also cost-effective and precise. It is recommended as an early diagnostic tool for CMT.